ウステキヌマブとは
開発の経緯
ウステキヌマブは、ヒトサイトカインのインターロイキン(IL)-12及びIL-23に共通するp40タンパク質サブユニットに特異的に結合する遺伝子組換えヒト免疫グロブリンG1κ(IgG1κ)モノクローナル抗体である。尋常性乾癬、乾癬性関節炎では、IL-12及びIL-23の異常発現が認められている。ウステキヌマブは、IL-12及びIL-23に共通するp40タンパク質サブユニットに結合することにより、IL-12及びIL-23がナチュラルキラー(NK)細胞又はT細胞表面のIL-12及びIL-23受容体複合体に結合することを阻害することによって、これらの疾患に対する作用を発揮すると考えられる。
ウステキヌマブBS皮下注45mgシリンジ「F」(以下、本剤)は「既存治療で効果不十分な下記疾患 尋常性乾癬、乾癬性関節炎 中等症から重症の活動期クローン病の維持療法(既存治療で効果不十分な場合に限る) 中等症から重症の潰瘍性大腸炎の維持療法(既存治療で効果不十分な場合に限る)」を効能又は効果とする先行バイオ医薬品ステラーラ®皮下注45mgシリンジ(一般名:ウステキヌマブ[遺伝子組換え])のバイオ後続品としてAlvotech社により開発された。本邦では富士製薬工業株式会社が、Alvotech社との独占的パートナーシップの合意に基づき、「バイオ後続品の品質・安全性・有効性確保のための指針」1)を踏まえた品質特性に関する試験及び非臨床試験を実施し、本剤と先行バイオ医薬品(EU、US及びJP)※との同等性・同質性を検討した。また、健康成人を対象とした海外第I相臨床試験(AVT04-GL-101試験)及び中等症又は重症の尋常性乾癬患者を対象とした海外第III相臨床試験(AVT04-GL-301試験)を実施し、本剤と先行バイオ医薬品(EU)との同等性を検討した。
富士製薬工業株式会社は、品質、非臨床、臨床のそれぞれの試験によって得られたエビデンスを評価した結果、本剤と先行バイオ医薬品(JP)との同等性・同質性が示されたことから、特許期間及び再審査期間が満了している「既存治療で効果不十分な下記疾患 尋常性乾癬、乾癬性関節炎」を本剤の効能又は効果として2023年9月に承認を取得した。
また、医療現場における利便性向上を鑑み、ウステキヌマブBS皮下注90mgシリンジ「F」の承認を2026年1月に取得した。
※先行バイオ医薬品(EU)は、欧州で承認されているステラーラ®〔ウステキヌマブ(遺伝子組換え)製剤〕を指す。
先行バイオ医薬品(US)は、米国で承認されているステラーラ®〔ウステキヌマブ(遺伝子組換え)製剤〕を指す。
先行バイオ医薬品(JP)は、本邦で承認されているステラーラ®〔ウステキヌマブ(遺伝子組換え)製剤〕を指す。
【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の効能又は効果】
4.
効能又は効果 〇既存治療で効果不十分な下記疾患 尋常性乾癬、乾癬性関節炎
特性
製品の治療学的特性
1.本剤は、ウステキヌマブ(遺伝子組換え)のバイオ後続品(バイオシミラー)である。
2.中等症又は重症の尋常性乾癬患者を対象とした海外第III相臨床試験(AVT04-GL-301試験)により、先行バイオ医薬品(EU)※1との有効性における同等性が確認された(検証的解析結果)※2。
3.重大な副作用として、アナフィラキシー(頻度不明)、重篤な感染症(1~5%未満)、結核(頻度不明)、間質性肺炎(頻度不明)が報告されている※3。
なお、主な副作用として、鼻咽頭炎、上気道感染、頭痛、浮動性めまい、咽喉頭疼痛、悪心、嘔吐、発疹、そう痒症、関節痛、注射部位反応、疲労が報告されている。
詳細については電子添文の副作用及び臨床成績の安全性の結果を参照すること。
製品の製剤学的特性
1.IL-12及びIL-23のp40サブユニットに結合する遺伝子組換えヒト免疫グロブリンG1κモノクローナル抗体であるウステキヌマブとして日本初のバイオシミラーである。
2.穿刺防止安全装置付きのプレフィルドシリンジ製剤である。
3.45mgシリンジ製剤の他に同剤形・同濃度・含量違いの90mgシリンジ製剤がある。
※1 先行バイオ医薬品(EU)は、欧州で承認されているステラーラ®〔ウステキヌマブ(遺伝子組換え)製剤〕を指す。
※2 製品の治療学的特性2の臨床試験では、体重100kg超の患者に対し、国内未承認である、初回からウステキヌマブ90mg投与〔本剤45mg又は先行バイオ医薬品(EU)45mgを2本投与〕を推奨した。
※3 重大な副作用の発現頻度は先行バイオ医薬品(ステラーラ®)の臨床試験結果に基づく副作用発現頻度である。
【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の効能又は効果、用法及び用量】
4.
効能又は効果 〇既存治療で効果不十分な下記疾患 尋常性乾癬、乾癬性関節炎
6.
用法及び用量 通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
有効成分に関する理化学的知見
一般名:
ウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1](JAN)
Ustekinumab(Genetical Recombination)[Ustekinumab Biosimilar 1](JAN)
本 質:
ウステキヌマブ[ウステキヌマブ後続1](以下、ウステキヌマブ後続1)は、インターロイキン(IL)-12及びIL-23のp40サブユニットに対する遺伝子組換えモノクローナル抗体であり、ヒトIgG1に由来する。ウステキヌマブ後続1は、Sp2/0細胞により産生される。ウステキヌマブ後続1は、449個のアミノ酸残基からなるH鎖(γ1鎖)2本及び214個のアミノ酸残基からなるL鎖(κ鎖)2本で構成される糖タンパク質(分子量:約149,000)である。
分子式:
C6482H10004N1712O2016S46(タンパク質部分)
分子量:
約149,000
薬物動態
1.血清中濃度
(1)単回投与
1)日本人及び外国人健康成人への単回投与時の薬物動態(海外第I相臨床試験[AVT04-GL-101試験])2)(日本人及び外国人データ)
海外において、日本人を含む健康成人に本剤及び先行バイオ医薬品(EU)*をウステキヌマブ(遺伝子組換え)として45mgを単回皮下投与したときのPK解析対象〔本剤:96例、先行バイオ医薬品(EU):97例〕における薬物動態(PK)パラメータ、血清中濃度を示す。
このうち最高血清中濃度(Cmax)及び0時間(投与前)から最終定量可能濃度までの血清中濃度-時間曲線下面積(AUC0-t)の幾何最小二乗(LS)平均値比の90%信頼区間はいずれも生物学的同等性の基準範囲内(0.80~1.25)であった。
*先行バイオ医薬品(EU):欧州で承認されているステラーラ®〔ウステキヌマブ(遺伝子組換え)製剤〕を指す。
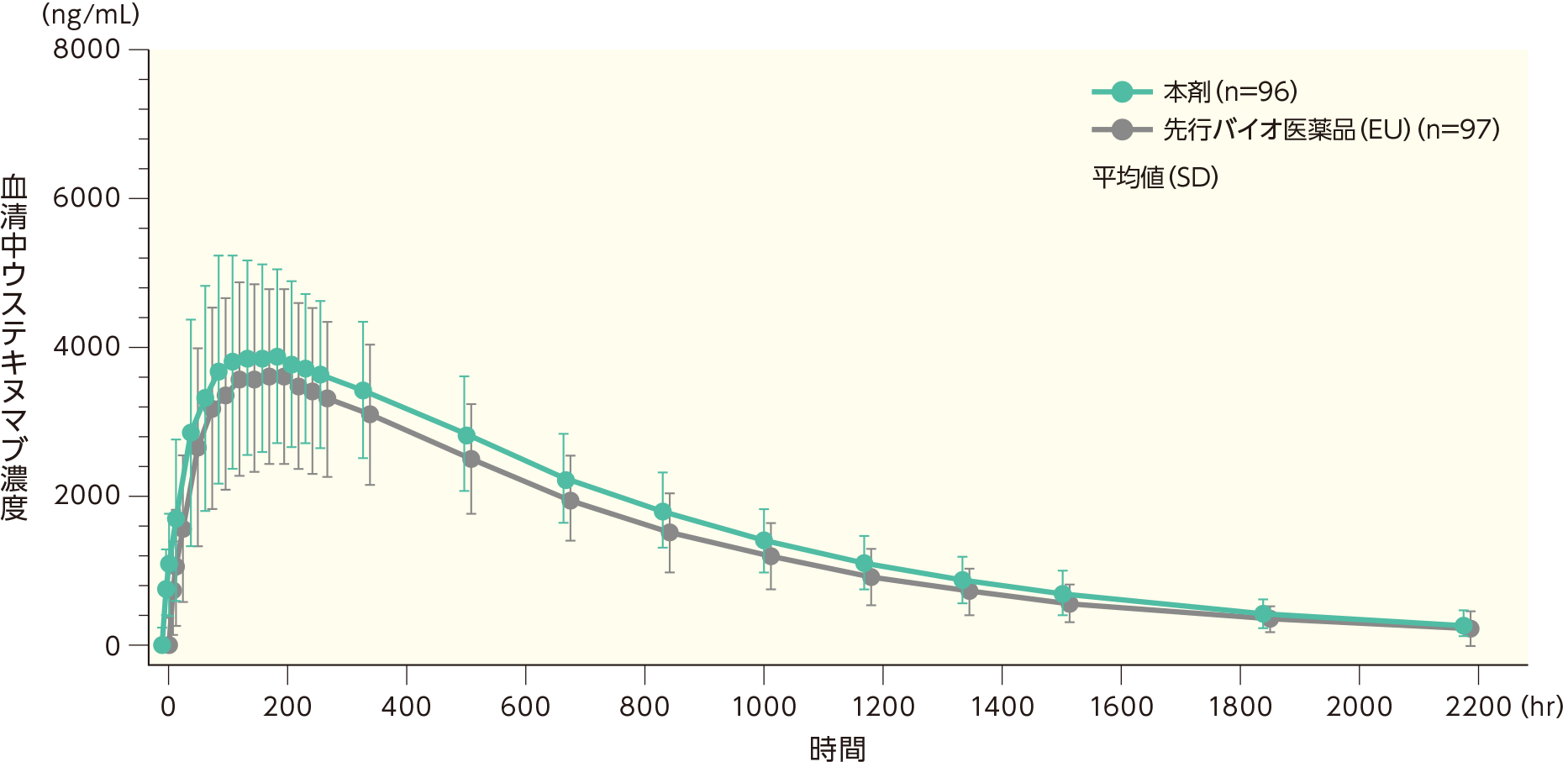
| Cmax | AUC0-t | AUC0-inf | Tmax | t1/2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | ng/mL | n | h·ng/mL | n | h·ng/mL | n | hr | n | hr | |
| 本剤 | 96 | 4,225 (1,348.22) | 96 | 3,432,850 (969,366) | 93 | 3,679,852 (1,066,729) | 96 | 180.22 (88.639) | 96 | 492.33 (124.766) |
| 先行バイオ 医薬品(EU) | 97 | 3,906 (1,247.47) | 97 | 3,044,207 (943,154) | 97 | 3,206,783 (1,029,191) | 97 | 175.12 (78.833) | 97 | 446.75 (109.936) |
平均値(SD)
AUC0-inf:0時間(投与前)から最終定量可能濃度に基づき無限大時間まで外挿した血清中濃度-時間曲線下面積、Tmax:最高血清中濃度到達時間、t1/2:消失半減期
2)日本人尋常性乾癬及び乾癬性関節炎患者への単回投与時の薬物動態
※ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」電子添文より
〈ステラーラ®皮下注45mgシリンジ〉※
乾癬患者に本剤22.5mg※、45mg及び90mgを単回皮下投与したとき、血清中ウステキヌマブ濃度は投与6.99~10.49日後に最高濃度に達した後、約3週の消失半減期で低下した。血清中ウステキヌマブのCmax及びAUC∞は、22.5~90mgの用量範囲において用量にほぼ比例して増加した3,4)。
薬物動態パラメータ
| 用量 | 22.5mg※ | 45mg | 90mg |
|---|---|---|---|
| Cmax(μg/mL) | 1.44(1.21~1.70) | 2.77(2.63~3.38) | 9.58(7.23~10.20) |
| Tmax(day) | 6.99(4.76~12.24) | 10.48(4.73~14.00) | 10.49(6.98~13.99) |
| AUC∞(μg·day/mL) | 61.3(49.2~75.8) | 109.4(96.9~171.9) | 242.7(195.7~272.3) |
中央値(四分位範囲)、各6例
※本剤の承認用量は1回45/90mgである。
3)日本人尋常性乾癬及び乾癬性関節炎患者への反復投与時の薬物動態
〈ステラーラ®皮下注45mgシリンジ〉※
乾癬患者に0、4週及びその後12週毎に52週目まで本剤45mg又は90mgを反復皮下投与したとき、血清中ウステキヌマブ濃度は投与開始28週目までに定常状態に達した。本剤45mg又は90mgを反復皮下投与したとき、定常状態における血清中ウステキヌマブのトラフ濃度の中央値はそれぞれ0.25~0.31及び0.55~0.76μg/mLであり、用量にほぼ比例して上昇した5,6)。
4)日本人尋常性乾癬及び乾癬性関節炎患者における薬物動態への体重の影響(外国人データ)
〈ステラーラ®皮下注45mgシリンジ〉※
乾癬患者において、体重100kg超の患者に本剤90mgを投与したときの血清中ウステキヌマブのトラフ濃度は体重100kg以下の患者に本剤45mgを投与したときと同程度であった(外国人データ)7)。
【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の効能又は効果、用法及び用量】
4.
効能又は効果 〇既存治療で効果不十分な下記疾患 尋常性乾癬、乾癬性関節炎
6.
用法及び用量 通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
2.吸収(外国人データ)
※ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」電子添文より
〈ステラーラ®皮下注45mgシリンジ〉※
乾癬患者に本剤を単回静脈内投与*(0.09、0.27、0.9、4.5mg/kg)又は単回皮下投与(0.27、0.675、1.35、 2.7mg/kg)したときの血清中ウステキヌマブ濃度を用いて算出した、ウステキヌマブを皮下投与したときの絶対的バイオアベイラビリティは約57.2%と推定された(外国人データ)8)。
*本剤は皮下注射により投与します。静脈内投与製剤は国内未承認です。
3.代謝
〈ステラーラ®皮下注45mgシリンジ〉※
ウステキヌマブは、ヒトIgG1由来の抗体であることから、他の免疫グロブリン9)と同様に代謝されると推察される。
薬効薬理
1.作用機序
※ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」電子添文より
〈ステラーラ®皮下注45mgシリンジ〉※
In vitro試験において、本剤はヒトインターロイキン(IL)-12及びIL-23を構成するp40たん白サブユニットに特異的かつ高い親和性で結合し10)、IL-12及びIL-23受容体複合体への結合を阻害した11)。
インターロイキン(IL)-23とIL-12はT細胞の活性化に重要な役割を果たすサイトカインである。IL-23はp19とp40の2つのサブユニットから構成され、IL-12はp35とp40の2つのサブユニットから構成される12)。
ウステキヌマブは、IL-23及びIL-12に共通するサブユニットであるp40に結合することによって、IL-17を産生するヘルパーT細胞(Th)17の増殖・維持及びIFN-γ等を産生するTh1細胞へのナイーブT細胞からの分化を抑制することで、尋常性乾癬、乾癬性関節炎に対する臨床効果を発揮すると考えられる12)。
ウステキヌマブの作用機序
![ウステキヌマブ「F」のヒトIL-12及びIL-23に対する作用機序[皮膚]](/img/about/zu_p32.png)
(イメージ図)
12)を参考に作図
2.非臨床試験
(1)p40、IL-12及びIL-23結合活性13)(in vitro)
本剤及び先行バイオ医薬品(EU及びUS)のp40、IL-12、IL-23に対する結合活性を以下に示す。
p40に対する相対結合活性
| p40相対結合作用(標準品に対する活性比) | |
|---|---|
| 本剤(n=8) | 96~111% |
| 先行バイオ医薬品(EU)*1(n=18) | 90~111% |
| 先行バイオ医薬品(US)*2(n=13) | 89~111% |
IL-12に対する相対結合活性
| IL-12相対結合作用(標準品に対する活性比) | |
|---|---|
| 本剤(n=8) | 99~102% |
| 先行バイオ医薬品(EU)*1(n=18) | 92~109% |
| 先行バイオ医薬品(US)*2(n=13) | 94~104% |
IL-23に対する相対結合活性
| IL-23相対結合作用(標準品に対する活性比) | |
|---|---|
| 本剤(n=8) | 97~104% |
| 先行バイオ医薬品(EU)*1(n=18) | 89~110% |
| 先行バイオ医薬品(US)*2(n=13) | 82~110% |
(2)IL-12介在性のINF-γ放出に対する阻害活性14)(in vitro)
本剤及び先行バイオ医薬品(EU及びUS)のIL-12介在性のINF-γ放出阻害活性を以下に示す。
IL-12中和活性(NK92細胞からのIFN-γ放出抑制作用)
| IL-12中和作用(標準品に対する活性比) | |
|---|---|
| 本剤(n=8) | 98~117% |
| 先行バイオ医薬品(EU)*1(n=17) | 78~122% |
| 先行バイオ医薬品(US)*2(n=13) | 80~120% |
*1 先行バイオ医薬品(EU):欧州で承認されているステラーラ®〔ウステキヌマブ(遺伝子組換え)製剤〕を指す。
*2 先行バイオ医薬品(US):米国で承認されているステラーラ®〔ウステキヌマブ(遺伝子組換え)製剤〕を指す。
(3)ヒトIL-12及びIL-23の生理活性抑制(in vitro)
※ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」電子添文より
〈ステラーラ®皮下注45mgシリンジ〉※
In vitro試験において、IL-12及びIL-23によって活性化されるヘルパーT細胞及びナチュラルキラー細胞などの免疫担当細胞の細胞内シグナル伝達並びにIFN-γ、IL-17A、IL-17F及びIL-22の分泌を抑制した15)。
安全性薬理試験及び毒性試験
1.安全性薬理試験
日本、欧州及び米国のバイオ後続品の開発に関して適用される規制ガイドラインに従い、安全性薬理試験は実施していない。
2.毒性試験
(1)反復投与毒性試験16)(カニクイザル)
カニクイザルに本剤(0、5、15、45mg/kg)又は先行バイオ医薬品(CHN)*(45mg/kg)を週1回、4週間反復皮下 投与した結果、明らかな毒性及び投与部位に刺激性は認められなかった。試験期間を通じて死亡は認められず、一般状態、心血管系、中枢神経系、呼吸器系、臓器・組織の肉眼的及び組織学的検査に関連した変化は認められなかった。本剤の無毒性量は45mg/kgであった。
(2)局所刺激性試験17)(カニクイザル)
1)単回皮下投与による局所刺激性
カニクイザルに本剤(0.9又は9mg/kg)又は先行バイオ医薬品(CHN)*(0.9又は9mg/kg)を単回皮下投与した 試験において、投与日の午後、投与翌日の午前及び午後、投与後2日の午前に投与部位を観察し、Draize法により紅斑及び浮腫を0(紅斑及び浮腫なし)から4(重度の紅斑及び浮腫)の範囲でスコア化した結果、試験期間を通じて投与部位の皮膚刺激は認められなかった。
2)反復皮下投与による局所刺激性
また、カニクイザルに本剤(5、15又は45mg/kg)又は先行バイオ医薬品(CHN)*(45mg/kg)を週1回4週間反復 皮下投与(製剤濃度はすべての群で90mg/mL)した試験において、投与部位の皮膚刺激は認められなかった。
*先行バイオ医薬品(CHN):中国で承認されているステラーラ®〔ウステキヌマブ(遺伝子組換え)製剤〕を指す。
1)厚生労働省: バイオ後続品の品質・安全性・有効性確保のための指針(薬生薬審発0204第1号令和2年2月4日), 2020
2)社内資料: 海外第I相臨床試験成績(AVT04-GL-101)[承認時評価資料]
3)臨床薬理に関する概括評価(ステラーラ皮下注シリンジ: 2011年1月21日承認、CTD2.5.3.2)
4)国内第I相臨床試験(ステラーラ皮下注シリンジ: 2011年1月21日承認、CTD2.7.6.2)
5)国内第II/III相臨床試験成績(JNS009-JPN-02)(ステラーラ皮下注シリンジ: 2011年1月21日承認、CTD2.7.6.7)
6)反復皮下投与試験(ステラーラ皮下注シリンジ: 2011年1月21日承認、審査報告書)
7)臨床薬理に関する概括評価(ステラーラ皮下注シリンジ: 2011年1月21日承認、CTD2.5.3.3)
8)海外臨床試験(ステラーラ皮下注シリンジ: 2011年1月21日承認、審査報告書)
9)Tabrizi MA, et al.: Drug Discov Today. 2006; 11(1-2): 81-88
10)Luo J, et al.: J Mol Biol. 2010; 402(5): 797-812
11)ウステキヌマブのヒトIL-12及びIL23中和作用機序(ステラーラ皮下注シリンジ: 2011年1月21日承認、CTD2.6.2.1)
12)藤田英樹 他: 最新医学. 2014; 69(2): 244-249
13)社内資料: 薬理作用に関連する試験-p40、IL-12及びIL-23に対する結合活性-[承認時評価資料]
14)社内資料: 薬理作用に関連する試験-効力-[承認時評価資料]
15)ウステキヌマブのヒトIL-12及びIL-23中和作用の機能的効果(ステラーラ皮下注シリンジ: 2011年1月21日承認、CTD2.6.2.2)
16)社内資料: 反復投与毒性試験[承認時評価資料]
17)社内資料: 局所刺激性試験[承認時評価資料]
