
臨床成績
ウステキヌマブBS皮下注45mgシリンジ「F」(以下、本剤)は、承認外の効能又は効果、用法及び用量による臨床試験の成績も含めた臨床データパッケージで評価され承認された。そのため、国内で承認されている効能又は効果、用法及び用量と異なるデータも紹介している。
1. 臨床データパッケージ
| 試験の種類・ 相試験番号 | 試験国 | 対象・例数 | 試験目的 | 試験デザイン |
|---|---|---|---|---|
| 海外第I相 AVT04-GL-101 試験 | オーストラリア、ニュージーランド | 健康成人298例 (日本人20例) | 本剤45mg、先行バイオ医薬品(EU)*145mgのPKの同等性の検証、並びに安全性及び免疫原性を評価する | 無作為化、二重盲検、単回投与、並行群間比較試験 |
| 海外第III相 AVT04-GL-301 試験 | エストニア、ジョージア、ポーランド、ウクライナ | 中等症又は重症の 慢性尋常性乾癬患者 581例 | 本剤45mg又は90mg*2、先行バイオ医薬品(EU)*145mg又は90mg*2皮下投与時の有効性の同等性の検証、並びに安全性及び免疫原性を評価する | 無作為化、二重盲検、多施設共同試験 |
*1 先行バイオ医薬品(EU)は、欧州で承認されているステラーラ®〔ウステキヌマブ(遺伝子組換え)製剤〕を指す。
*2 海外第III相臨床試験(AVT04-GL-301試験)では、体重100kg超の患者に対しては国内未承認である、初回からウステキヌマブ90mg投与〔本剤45mg又は先行バイオ医薬品(EU)45mgを2本投与〕を推奨した。
【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の効能又は効果、用法及び用量】
4.
効能又は効果 〇既存治療で効果不十分な下記疾患 尋常性乾癬、乾癬性関節炎
6.
用法及び用量 通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
本試験には承認外の用法及び用量の成績が含まれているが、本剤と先行バイオ医薬品(EU)*1との有効性の同等性を検討した承認時評価資料のため紹介している。
2.海外第III相臨床試験(AVT04-GL-301試験)1,2)(海外データ)
1)社内資料: 海外第III相臨床試験成績(AVT04-GL-301)[承認時評価資料]
2)Feldman SR, et al.: Expert Opin Biol Ther. 2023; 23(8): 759-771
(COI:本試験はAlvotech社の資金提供により実施された。著者の中にAlvotech社の社員が含まれる)
| 目的 | 中等症又は重症の慢性尋常性乾癬患者を対象に、本剤と先行バイオ医薬品(EU)の有効性、安全性及び免疫原性を比較する。 |
|---|---|
| 試験デザイン | 無作為化、二重盲検、多施設共同試験 |
| 対象 | 中等症又は重症の慢性尋常性乾癬患者581例 |
| 試験方法 | 本試験はステージ1(初回投与から16週目まで)及びステージ2(16週目から52週目まで)で構成された。対象を、尋常性乾癬に対する生物学的製剤による治療歴の有無及び体重(80kg以下、80kg超-100kg以下、100kg超)を層別因子として層別し、本剤群又は先行バイオ医薬品群のいずれかに1:2の割合で無作為割り付けした。 本剤群:本剤の初回皮下投与*2を実施し、4週後に2回目の皮下投与*2を行った。その後は、以降40週まで12週ごとに皮下投与*2した。 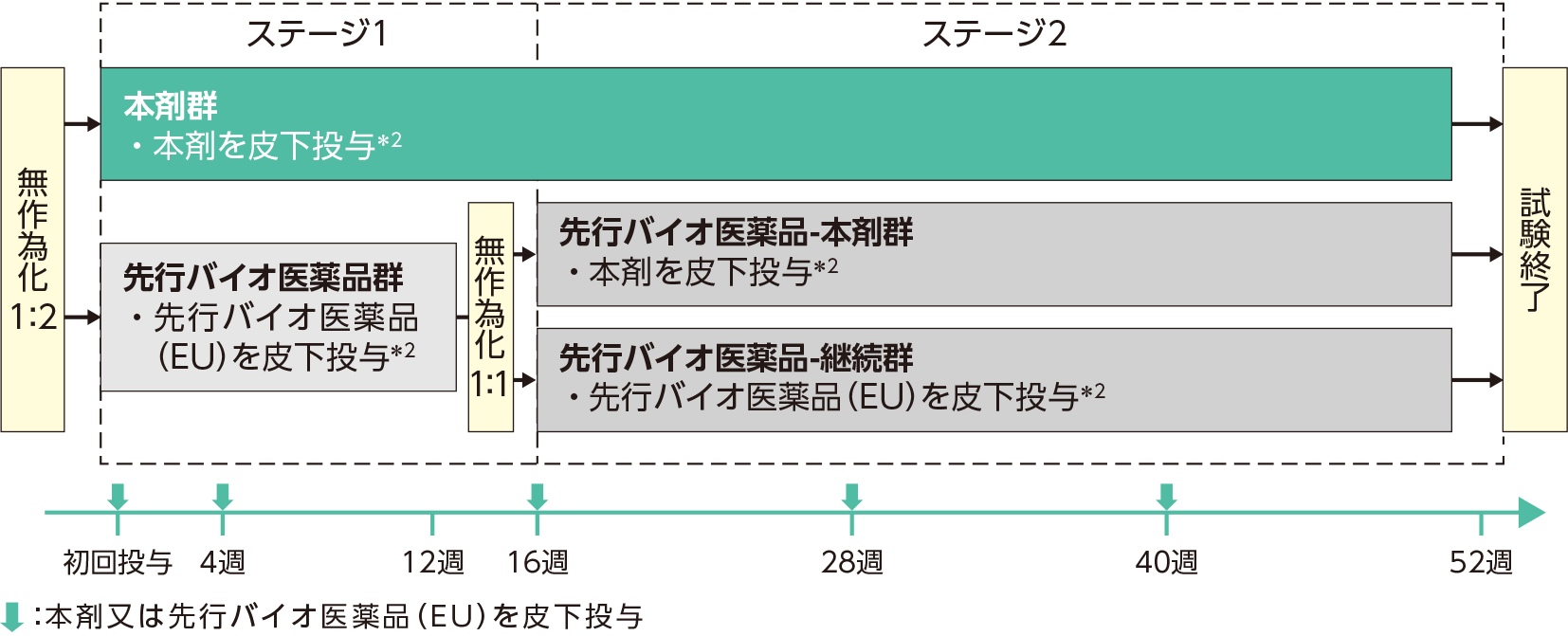 |
| 主要評価項目 | [有効性] ベースラインから12週時点までのPASI改善率[検証的解析項目] |
| 副次評価項目 | [有効性]
[安全性]
[免疫原性]
[薬物動態(PK)]
|
| 解析計画 | [有効性の解析] 主要評価項目:解析はPPS集団*3のうち、体重100kg以下の患者を対象に実施した。解析は独立して実施し、多重性の調整は行わなかった。共分散分析(ANCOVA)モデルを用いて解析した。ANCOVAモデルでは、PASI改善率を応答変数として、無作為化された投与群及びベースラインの尋常性乾癬に対する生物学的製剤による治療歴(あり/なし)を層別変数の因子とした。ベースラインのPASIスコア及び体重も連続共変量として含めた。12週時点における投与群間の調整済み平均差の推定値をモデルから算出し、調整済み平均差の両側95%信頼区間(CI)も算出して同等性を検討した。本剤群と先行バイオ医薬品群のPASI改善率の調整済平均差の95%CIが(-15%、15%)内に収まっている場合、臨床的同等性が示されたとした。 副次評価項目:解析はITT集団*4を対象に、本剤群と先行バイオ医薬品群の有効性を解析し、記述的に要約した。ベースラインからのPASI改善率及びベースラインから12週時点までのPASIのAUECの解析には、主要評価項目の解析に用いたANCOVAモデルを適用した。なお、多重性の調整は実施しなかった。 [安全性の解析] 有害事象*5は、安全性解析対象集団*6を対象として解析し、記述的に要約した。 [免疫原性解析] ADA及びnAbの有無、ADA陽性の抗体価を投与群及び来院ごとに集計した。 [PKの解析] 血清中トラフ濃度を来院及び試験期間別に経時的に記述的に要約した。 |
| 試験期間 | 2021年6月3日(最初の患者の初回来院)~2022年10月11日(最後の患者の最終来院) |
*1 先行バイオ医薬品(EU)は、欧州で承認されているステラーラ®〔ウステキヌマブ(遺伝子組換え)製剤〕を指す。
*2 患者体重が100kg以下の場合は本剤又は先行バイオ医薬品(EU)45mg、100kg超の場合は本剤又は先行バイオ医薬品(EU)90mgを皮下投与※
※初回からウステキヌマブ90mg投与は国内未承認です。
*3 PPS集団:治験実施計画書からの逸脱がなく、投与期間を完了した患者
*4 ITT集団:無作為化後に割り付けられた治験薬を1回以上投与されたすべての患者
*5 各有害事象はMedDRA v25.1に基づく。
*6 安全性解析対象集団:無作為化後に割り付けられた治験薬を1回以上投与されたすべての患者
【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の用法及び用量】
6.
用法及び用量 通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
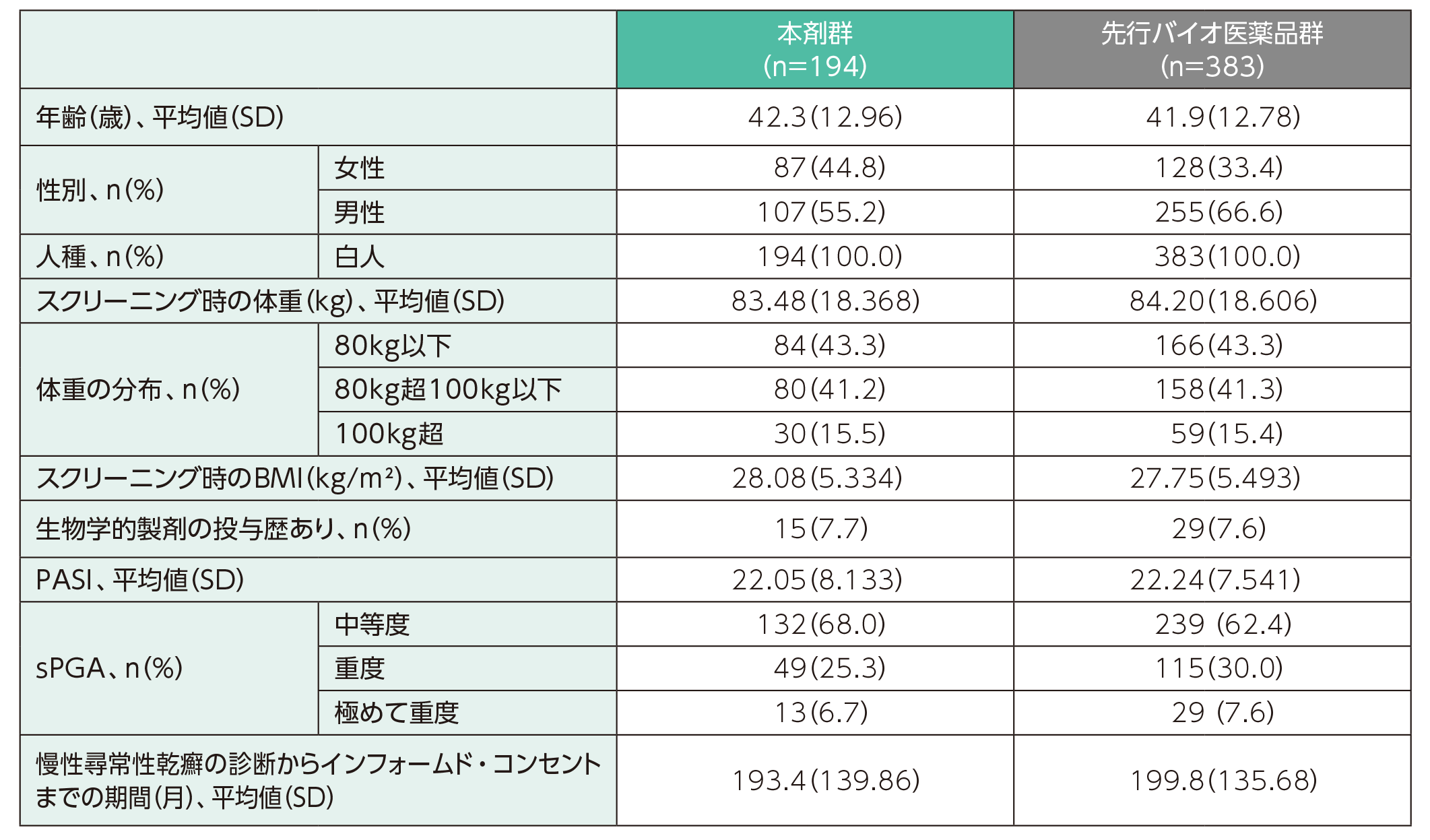
患者背景
人口統計学的特性(PPS集団、ベースラインから16週時点まで)ステージ1
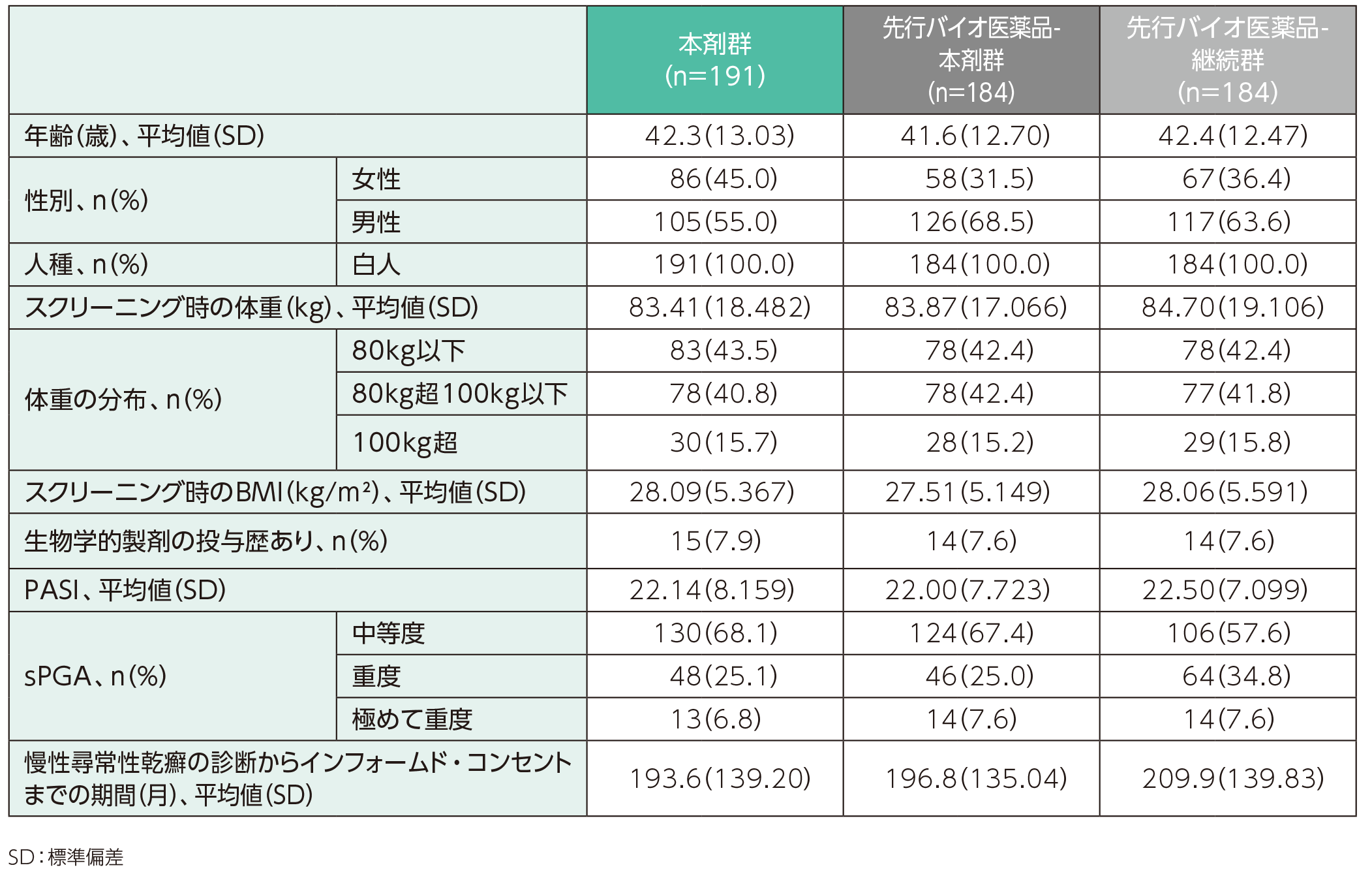
人口統計学的特性(ITT集団、ベースラインから52週時点まで)ステージ2
ベースラインから12週時点までのPASI改善率(主要評価項目)[検証的解析結果]ステージ1
(1)体重100kg以下の患者を対象とした解析
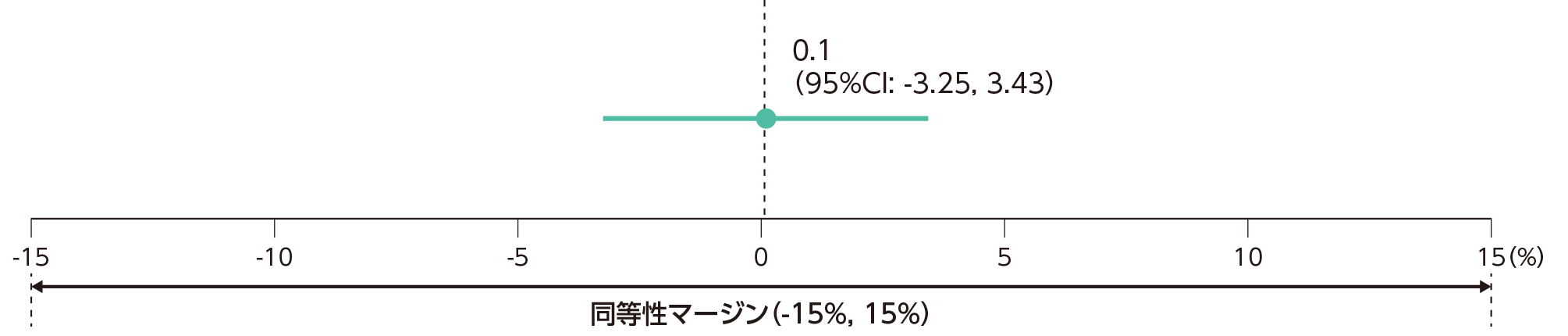
PPS集団のうち、体重100kg以下の患者集団におけるベースラインから12週時点までのPASI改善率のLS平均値(SE)は、本剤群で86.9(1.91)%、先行バイオ医薬品群で86.8(1.64)%であった。両群間の改善率のLS平均値の差(SE)は0.1(1.70)%であった。
両群間の改善率のLS平均値の差の95%CIは(-3.25%, 3.43%)であり、事前に規定した同等性マージン(-15%,15%)内であったことから、有効性の同等性が示された。
ベースラインから12週時点までのPASI改善率(PPS集団のうち、体重100kg以下の患者)
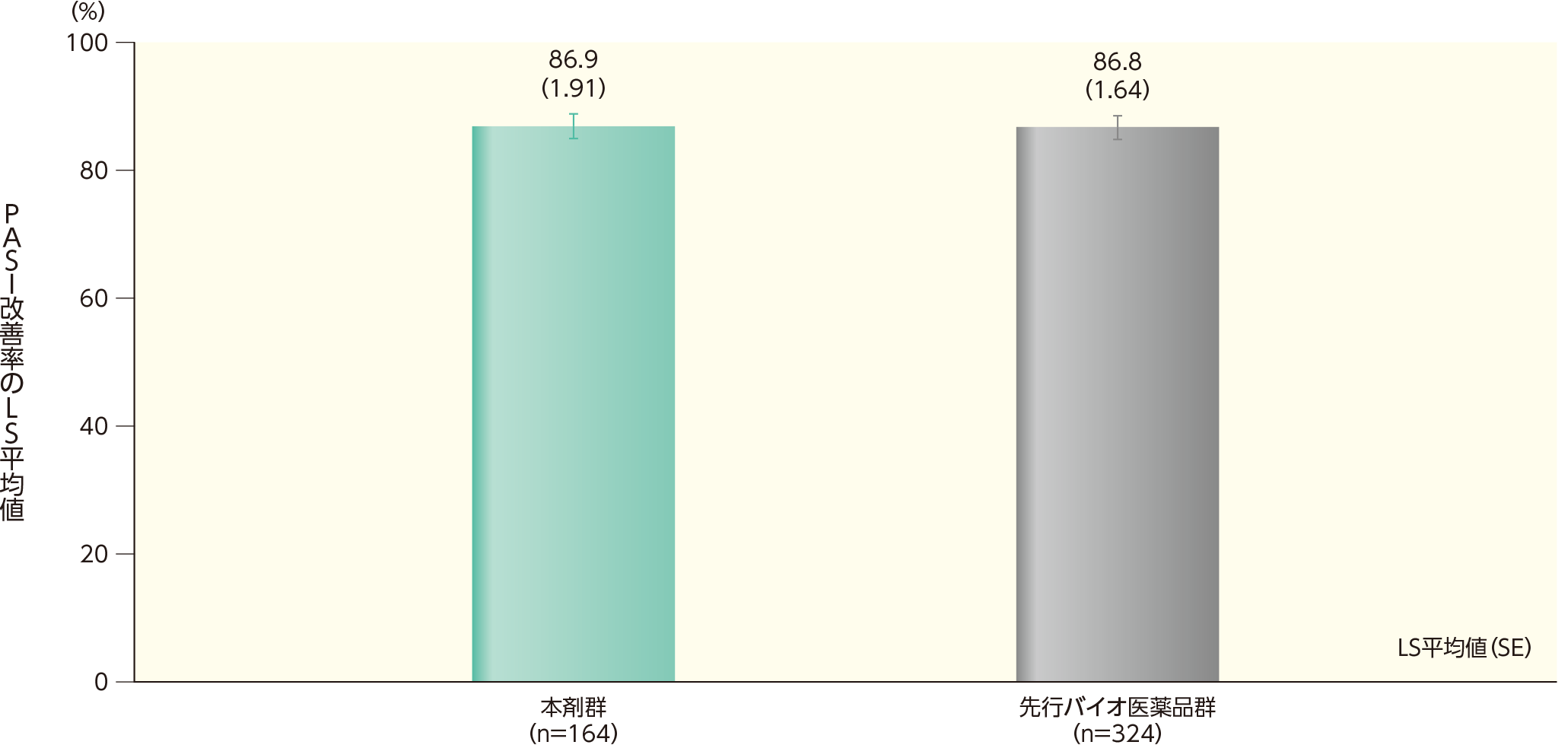

両群間の改善率のLS平均値の差(95%CI)(PPS集団のうち、体重100kg以下の患者)
(参考)PPS集団を対象とした解析ステージ1
PPS集団におけるベースラインから12週時点までのPASI改善率のLS平均値(SE)は、本剤群で87.3(1.73)%、先行バイオ医薬品群で86.8(1.49)%であった。両群間の改善率のLS平均値の差(SE)は0.4(1.56)%であった。
ベースラインから12週時点までのPASI改善率(PPS)

【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の用法及び用量】
6.
用法及び用量 通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
4、8、12、16、28、40、52週時点のPASI 50、75、90、100の奏効率(副次評価項目)
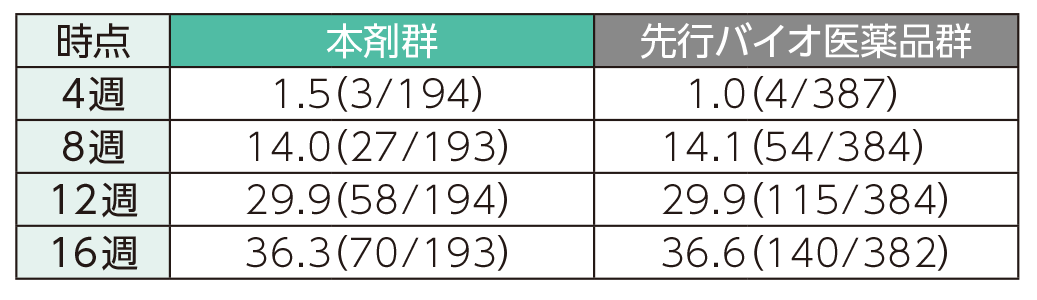
(1)4、8、12、16週時点のPASI 50、75、90、100の奏効率ステージ1
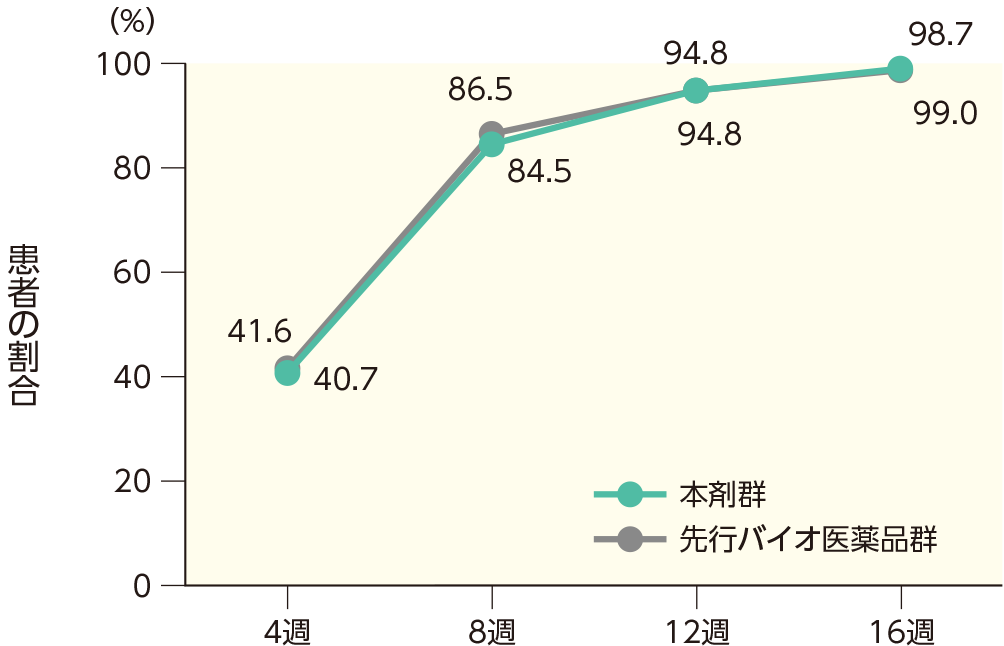
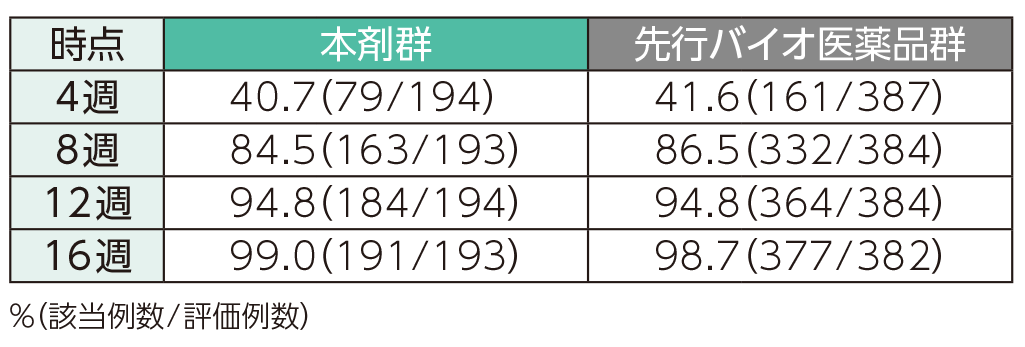
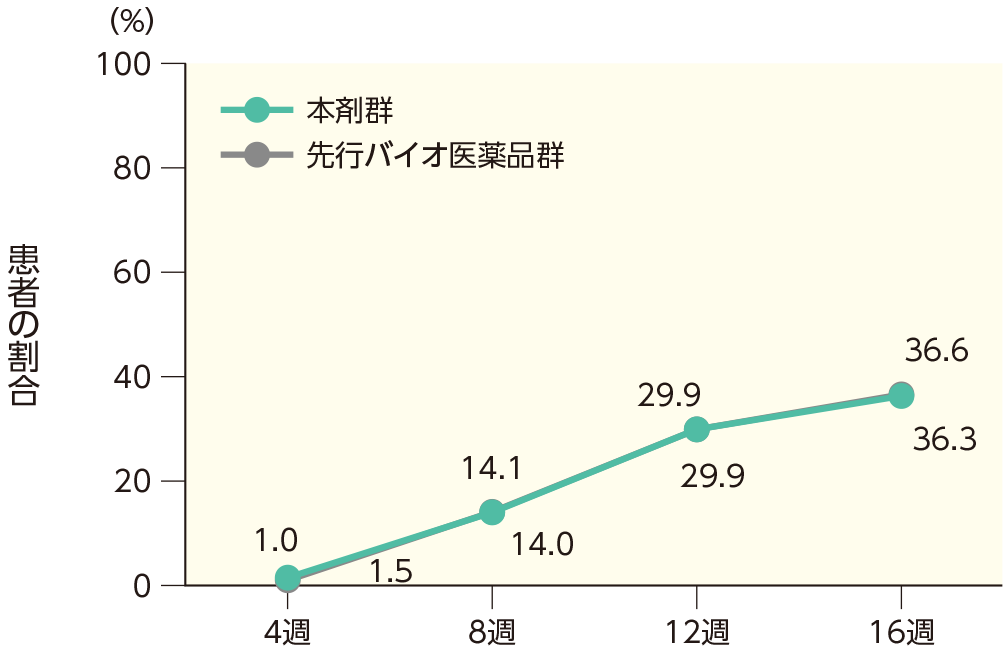
4週時点にPASI 50の奏効率を達成した患者の割合は、本剤群で40.7%、先行バイオ医薬品群で41.6%であった。各群で16週時点までにPASI 50、75、90、100の奏効率を達成した患者の割合は以下のとおりであった。
PASI 50の奏効率(ITT集団)
PASI 75の奏効率(ITT集団)
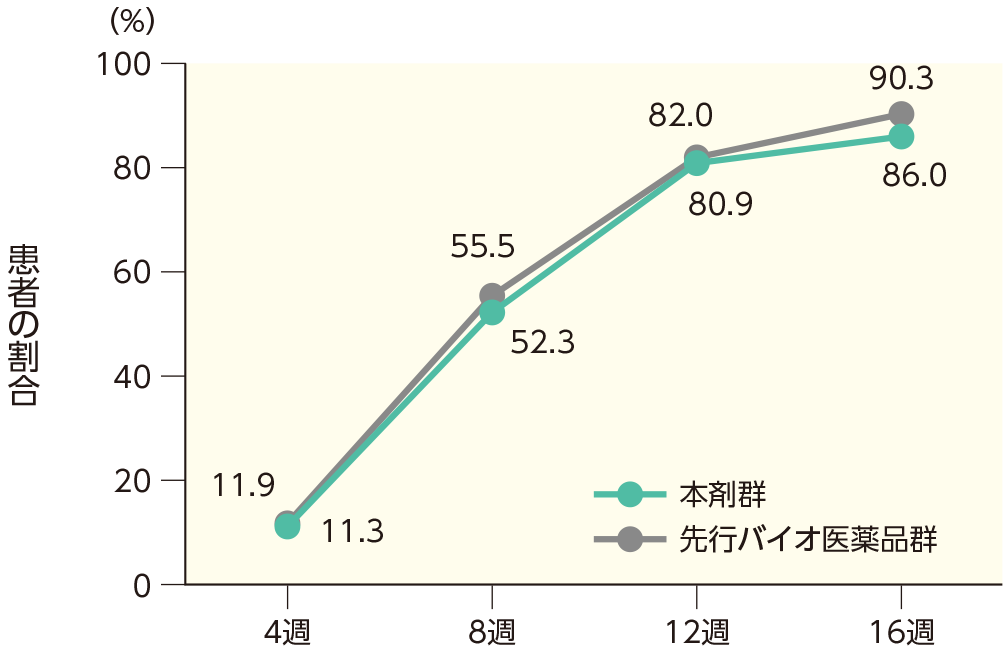
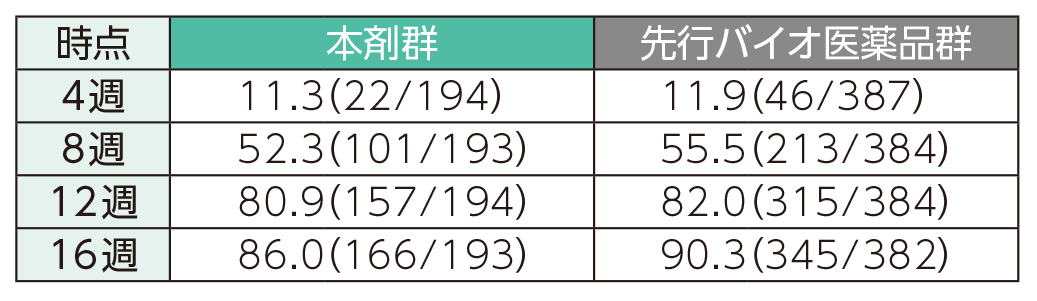
PASI 90の奏効率(ITT集団)
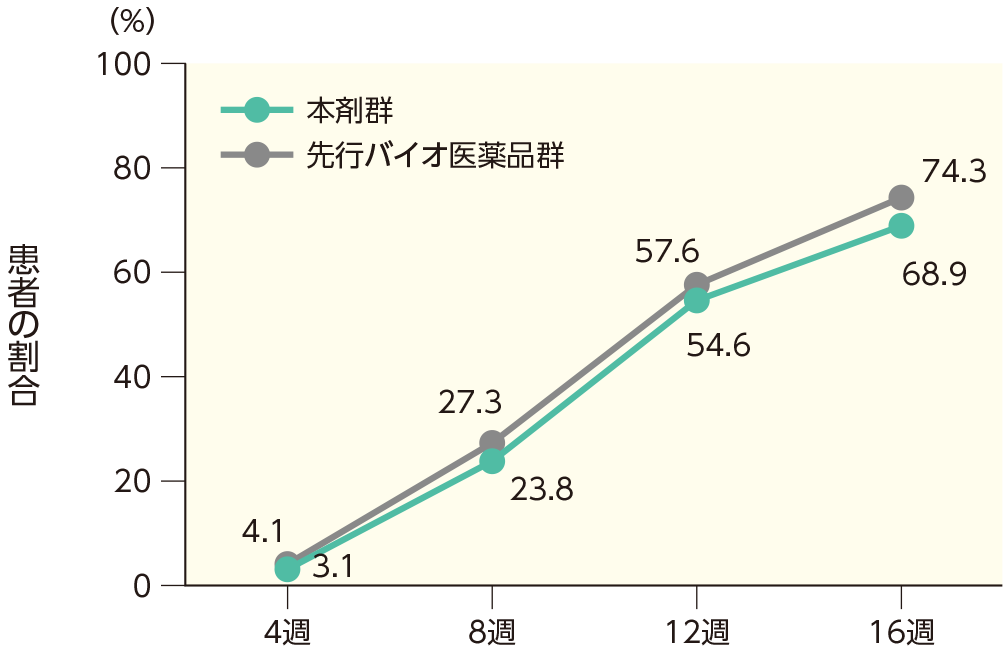
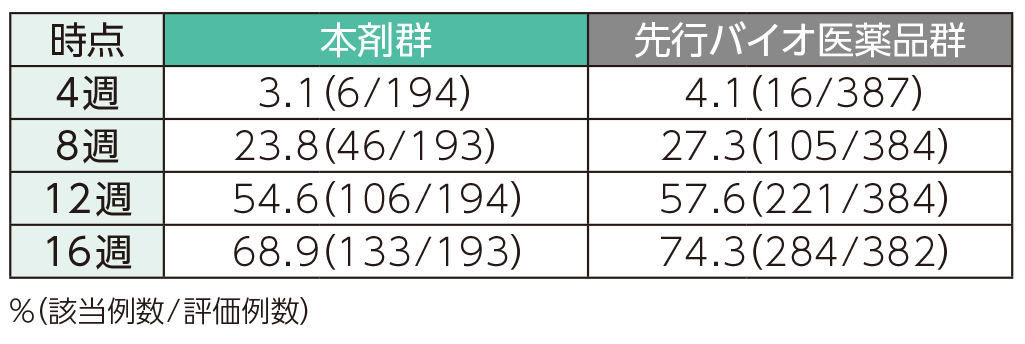
PASI 100の奏効率(ITT集団)
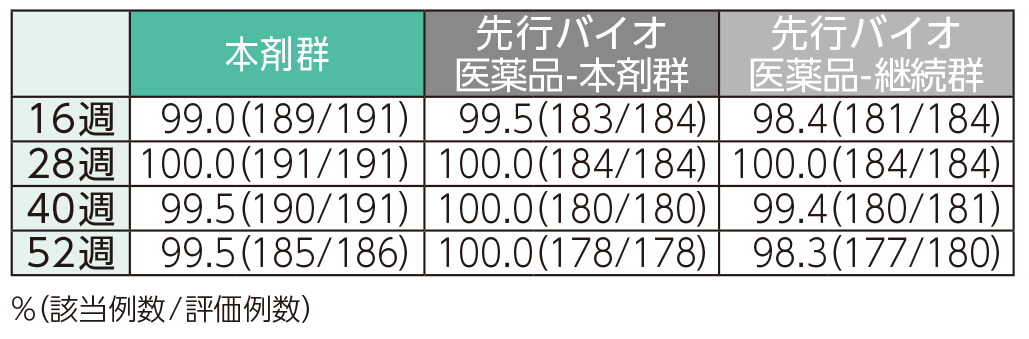
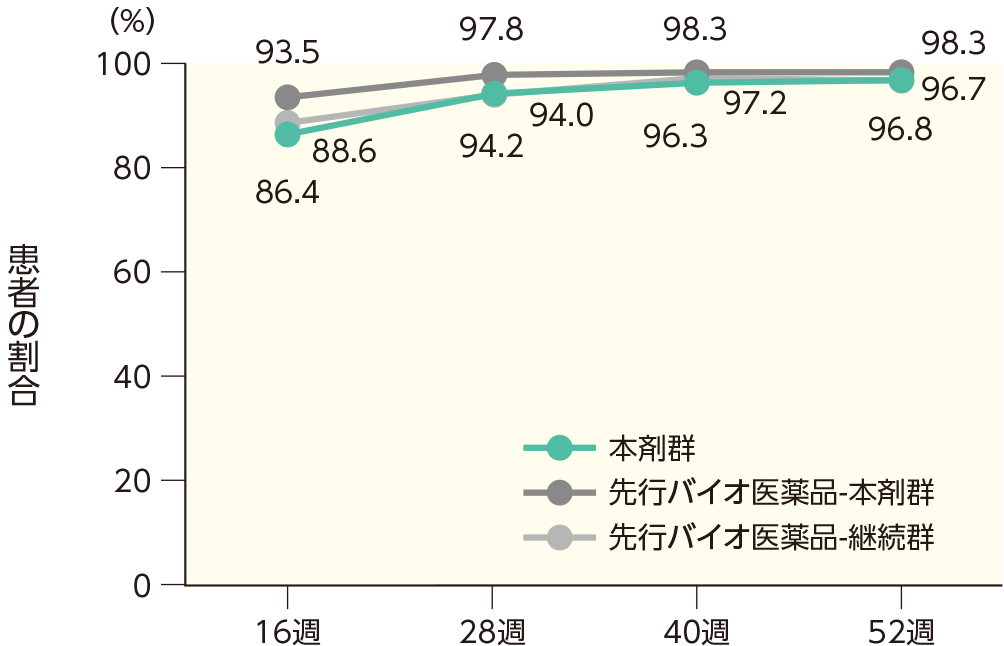
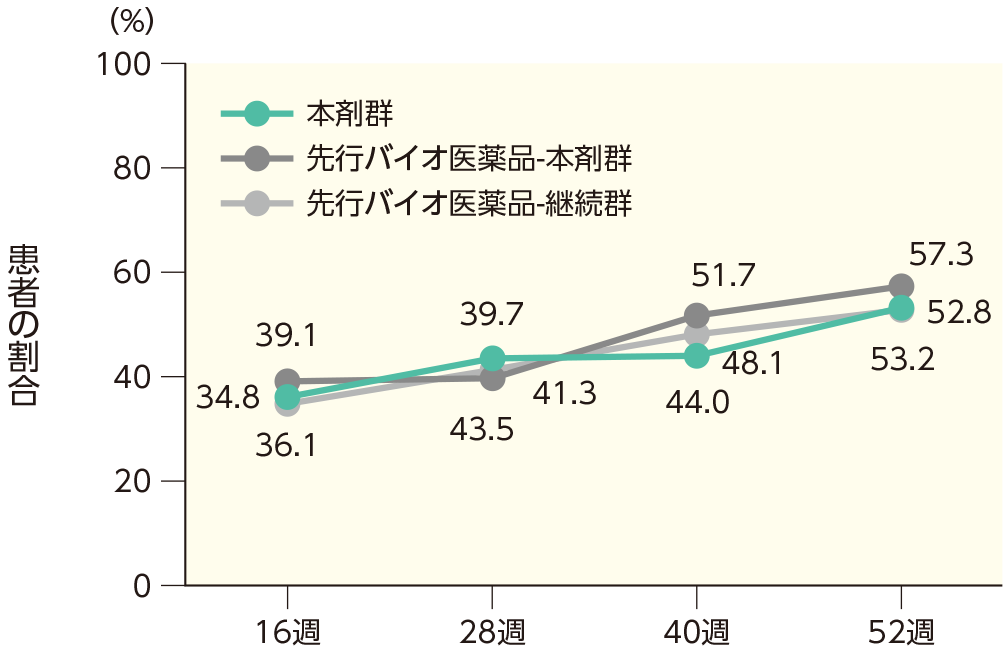
(2)16、28、40、52週時点のPASI 50、75、90、100の奏効率ステージ2
16週時点にPASI 50の奏効率を達成した患者の割合は、本剤群で99.0%、先行バイオ医薬品-本剤群で99.5%、先行バイオ医薬品-継続群で98.4%であった。各群で52週時点までにPASI 50、75、90、100の奏効率を達成した患者の割合は以下のとおりであった。
PASI 50の奏効率(ITT集団)

PASI 75の奏効率(ITT集団)

PASI 90の奏効率(ITT集団)
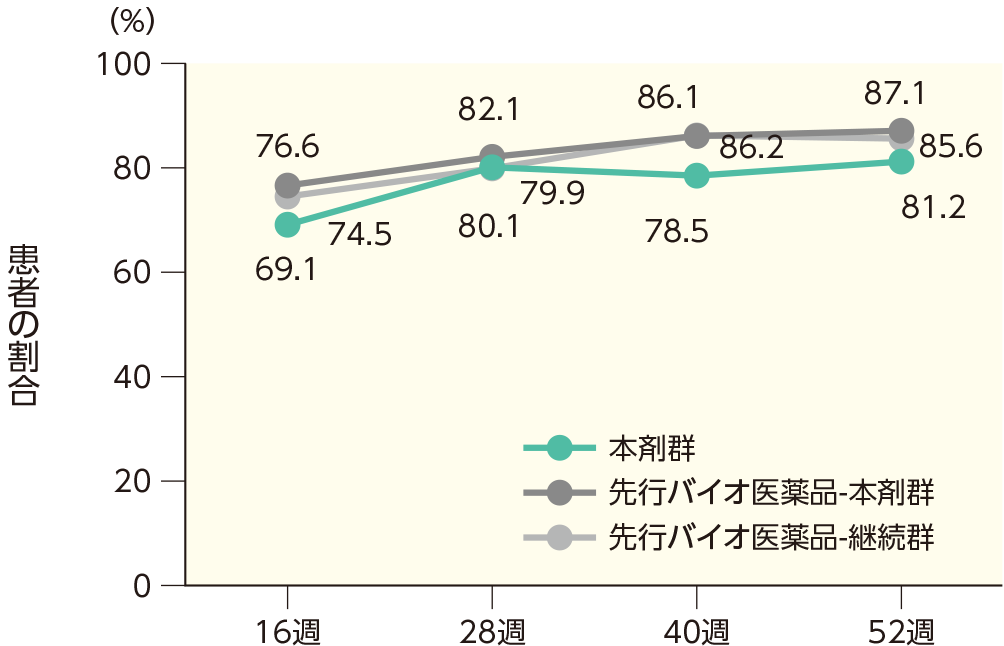

PASI 100の奏効率(ITT集団)

【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の用法及び用量】
6.
用法及び用量 通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
ベースラインから4、8、16、28、40、52週時点までのPASI改善率(副次評価項目)
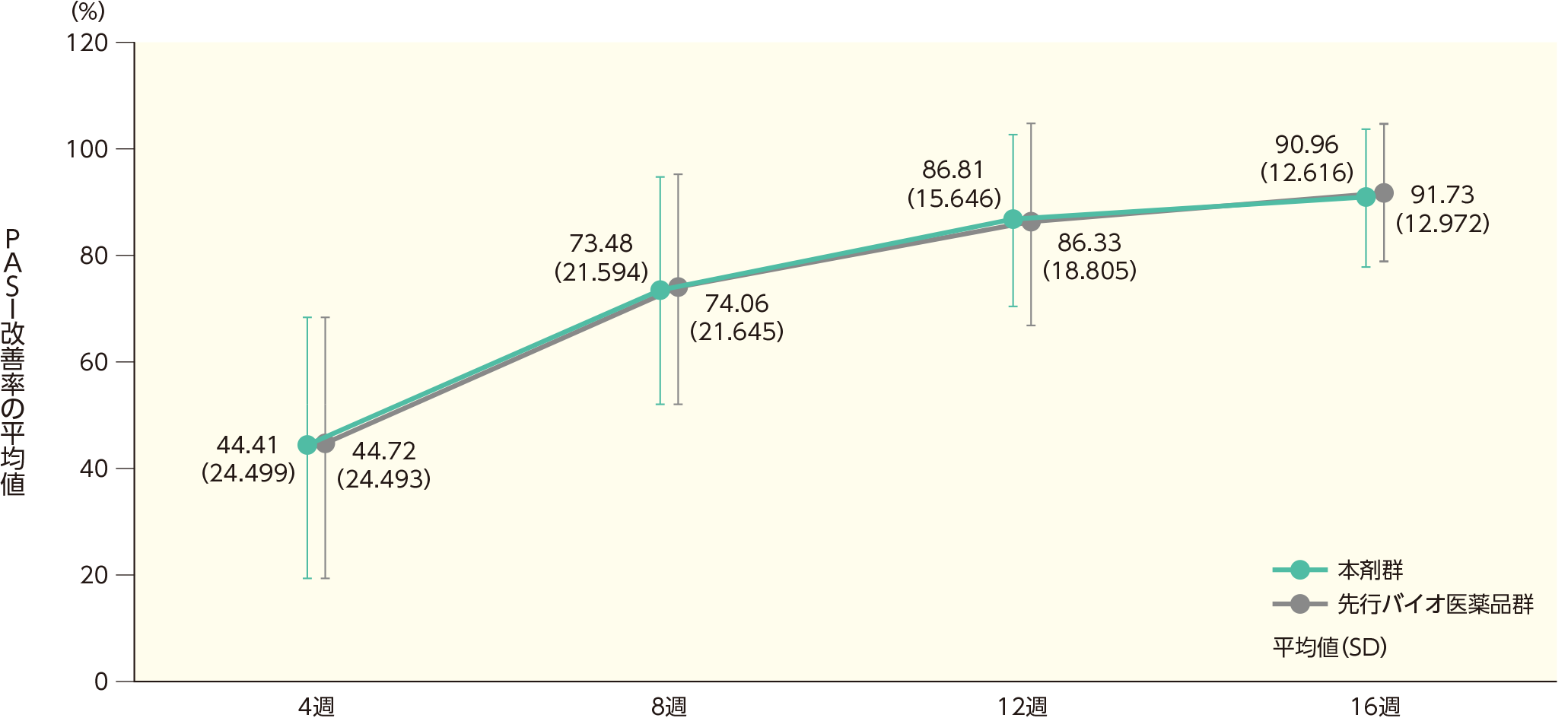
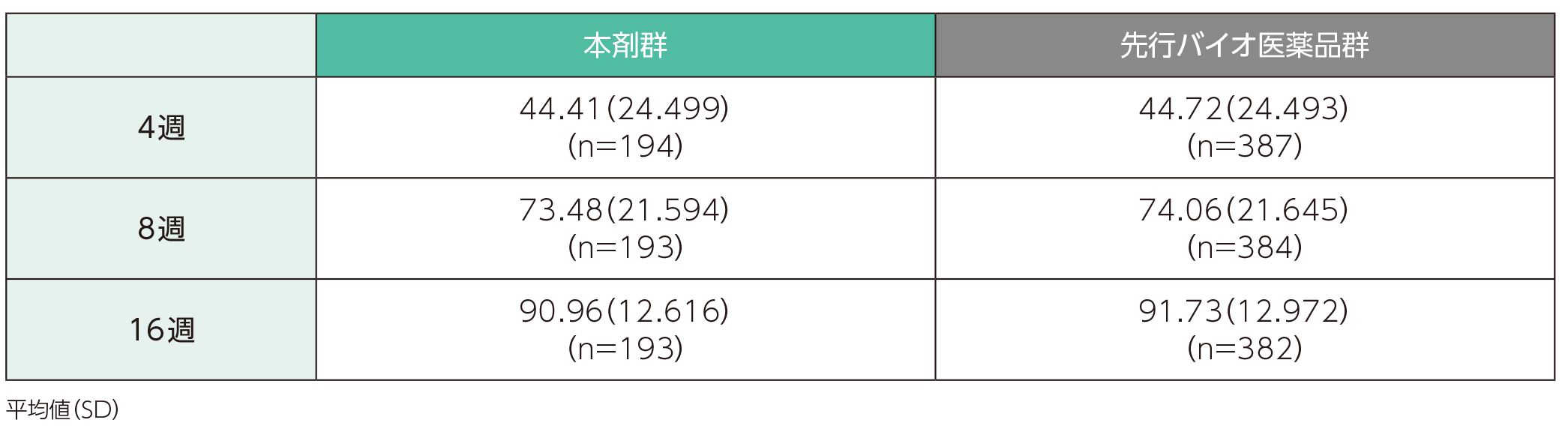
(1)ベースラインから4、8、16週時点までのPASI改善率ステージ1
ベースラインから4週時点までのPASI改善率の平均値(SD)は、本剤群で44.41(24.499)%、先行バイオ医薬品で44.72(24.493)%であった。各群の16週時点までのPASI改善率の平均値(SD)は以下のとおりであった。
ベースラインから4、8、16週時点までのPASI改善率(ITT集団)
【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の用法及び用量】
6.
用法及び用量 通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
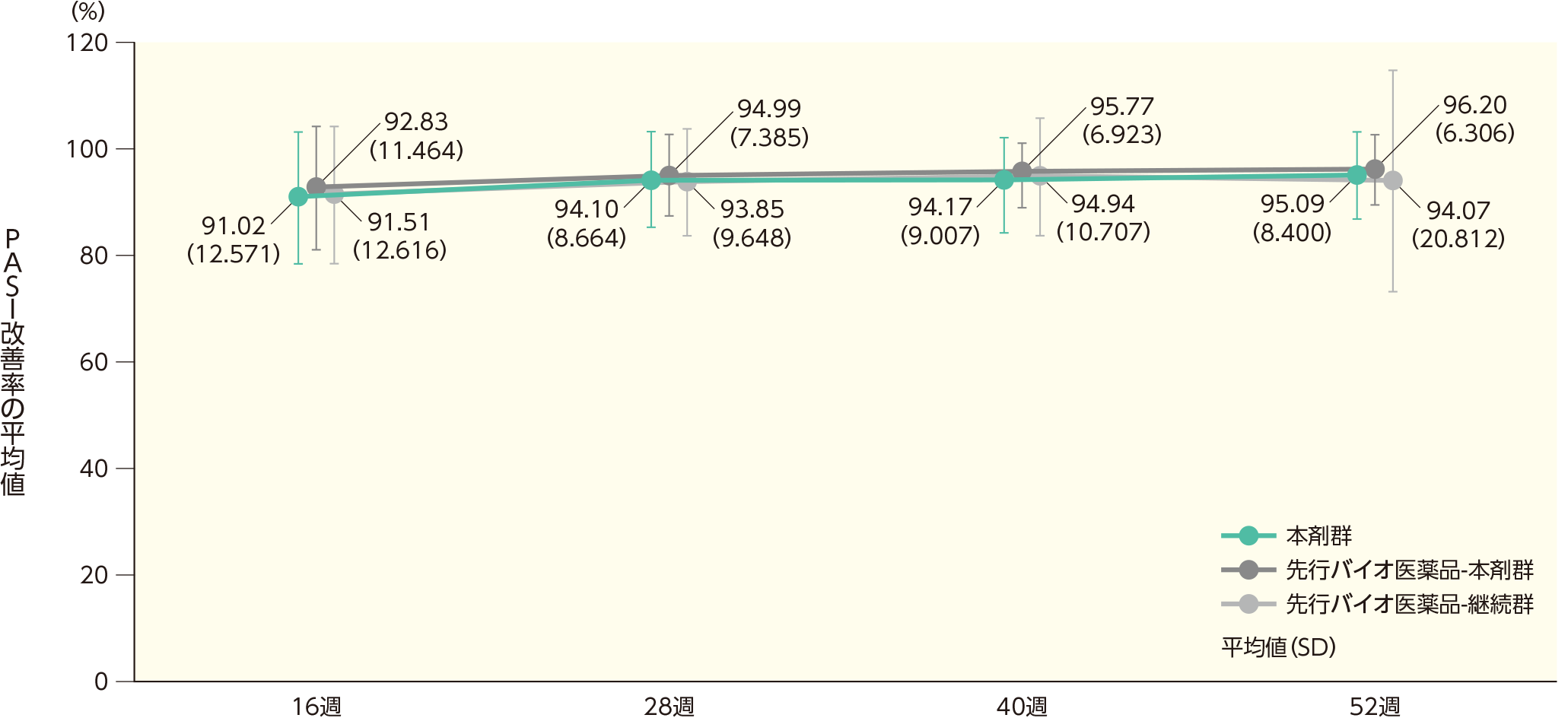
(2)ベースラインから16、28、40、52週時点までのPASI改善率ステージ2
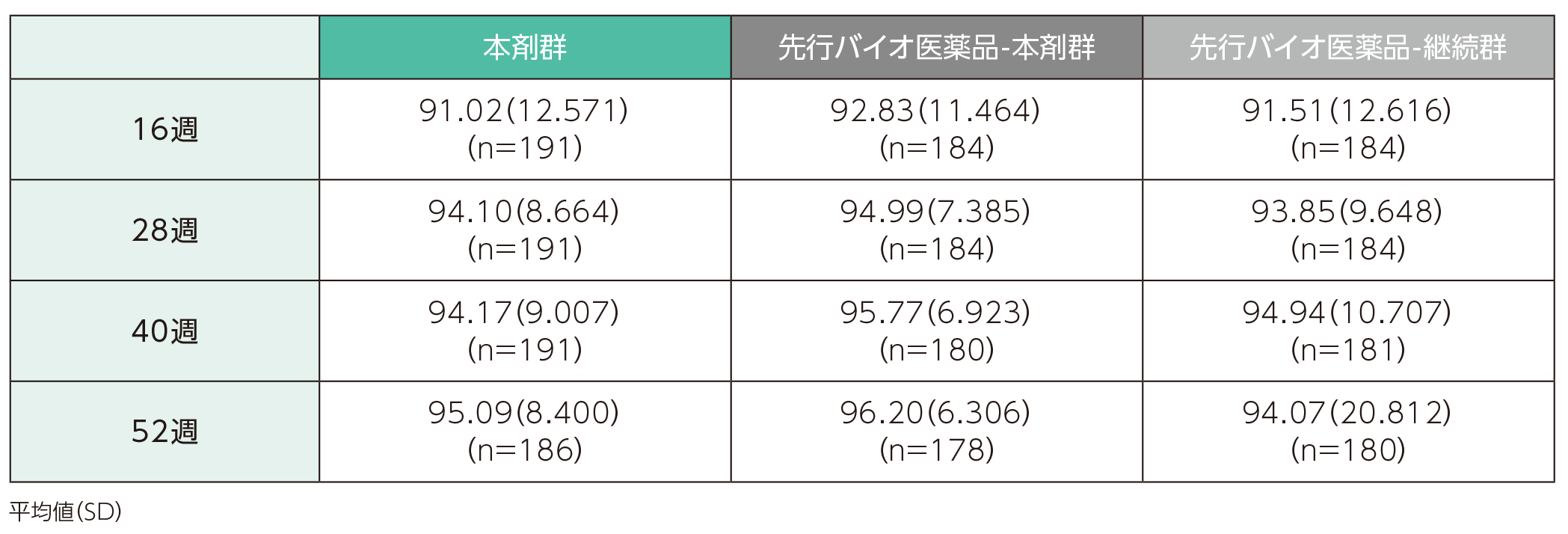
ベースラインから16週時点までのPASI改善率の平均値(SD)は、本剤群で91.02(12.571)%、先行バイオ医薬品-本剤群で92.83(11.464)%、先行バイオ医薬品-継続群で91.51(12.616)%であった。各群の52週時点までのPASI改善率の平均値(SD)は以下のとおりであった。
ベースラインから16、28、40、52週時点までのPASI改善率(ITT集団)
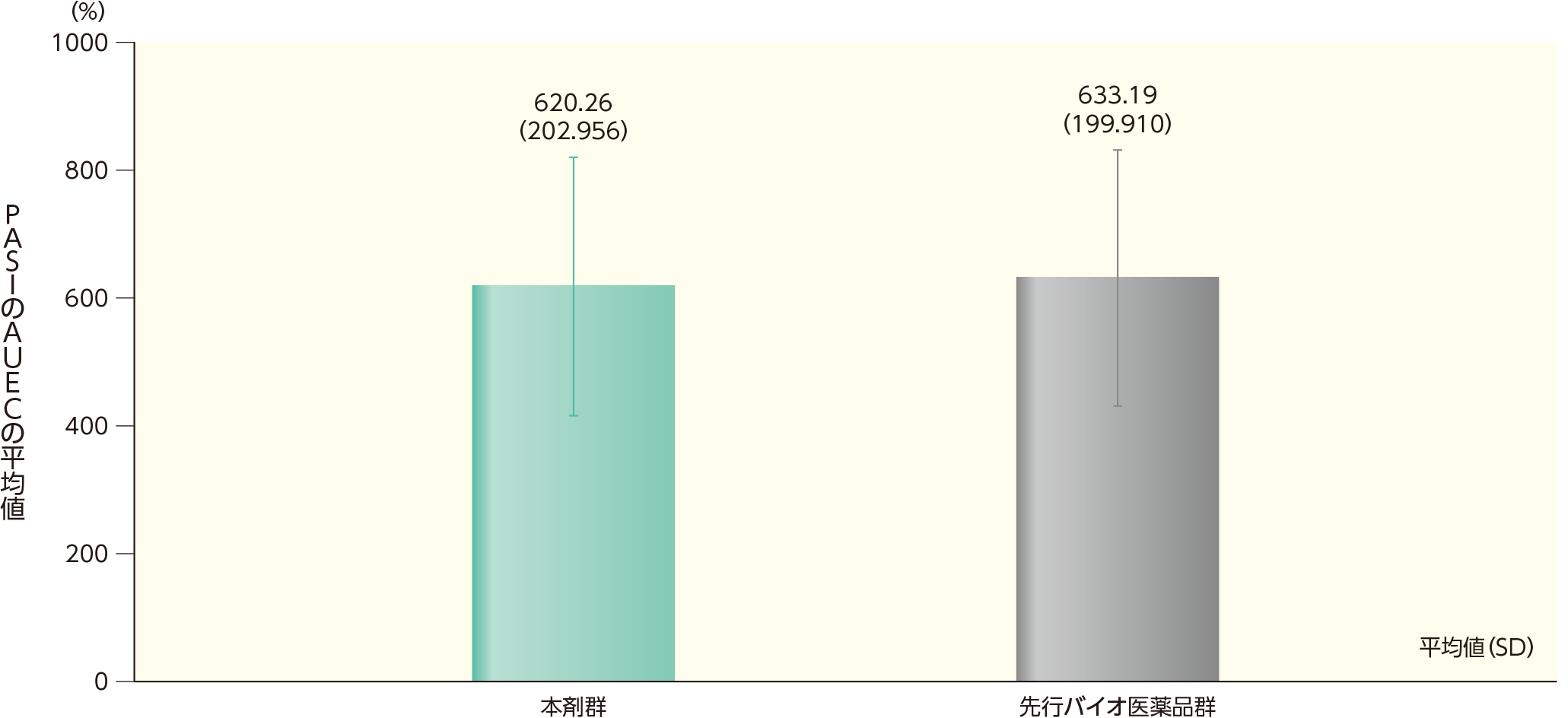
ベースラインから12週時点までのPASIの効果曲線下面積(AUEC) (副次評価項目)ステージ1
ベースラインから12週時点までのPASIのAUECの平均値(SD)は、本剤群で620.26(202.956)、先行バイオ医薬品群で633.19(199.910)であった。
ベースラインから12週時点までのPASIのAUEC(ITT集団)

【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の用法及び用量】
6.
用法及び用量 通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
4、8、12、16、28、40、52週時点に静的全般評価(sPGA)で「消失」又は「ほぼ消失」を達成した患者の割合(副次評価項目)
(1)4、8、12、16週時点にsPGAで「消失」又は「ほぼ消失」を達成した患者の割合ステージ1
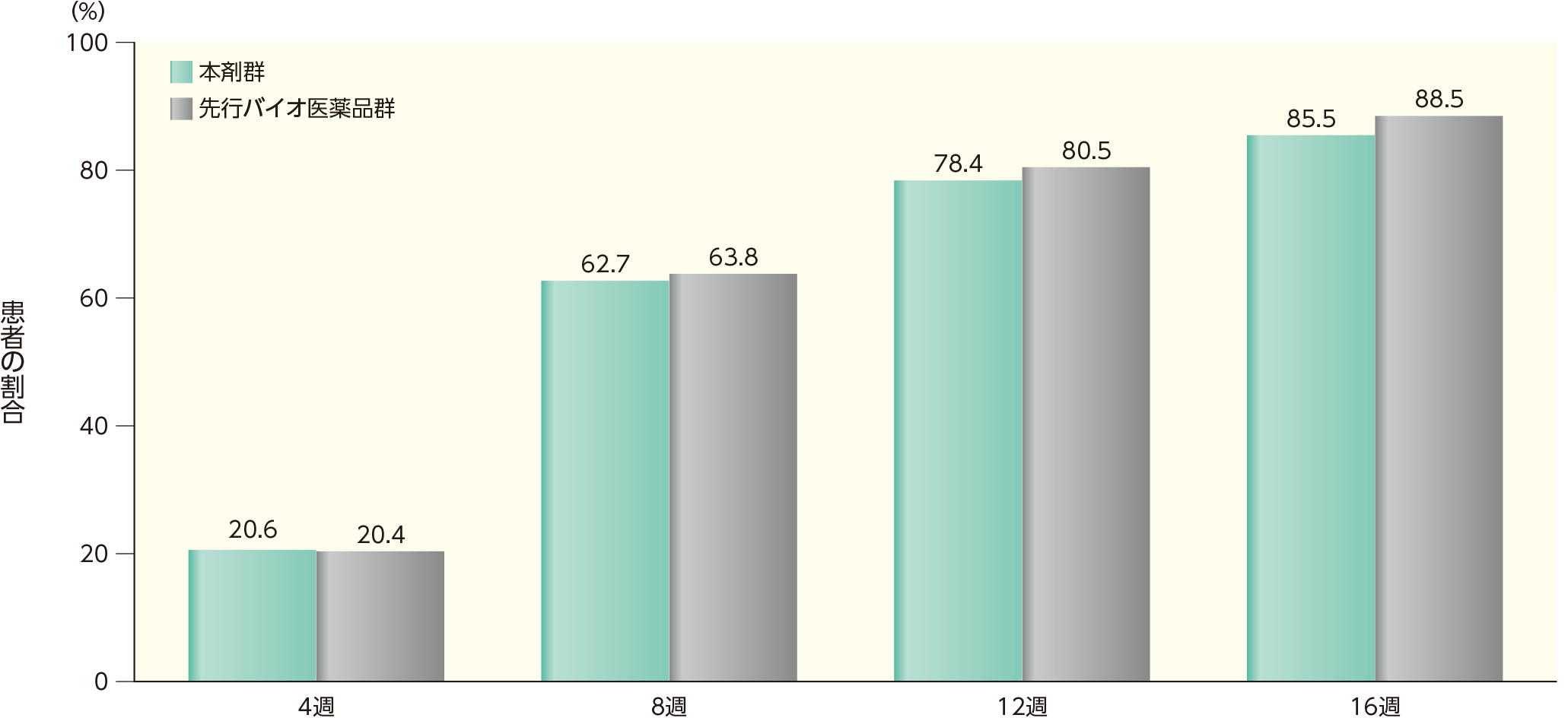
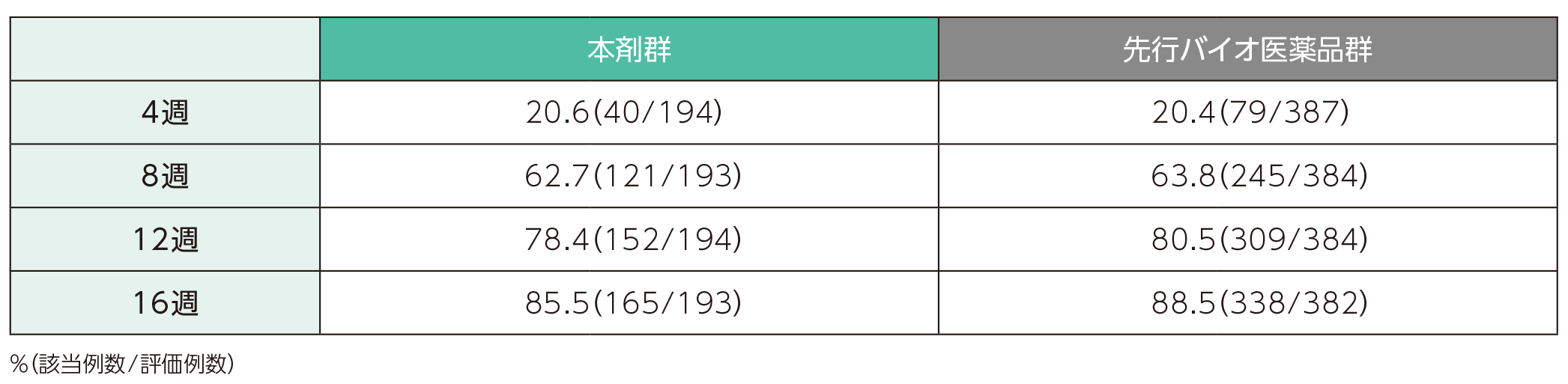
4週時点において、sPGAで「消失」又は「ほぼ消失」*を達成した患者の割合は、本剤群で20.6%、先行バイオ医薬品群で20.4%であった。各群で16週時点までにsPGAで「消失」又は「ほぼ消失」を達成した患者の割合は以下のとおりであった。
*sPGAスコアが0の場合を「消失」、1の場合を「ほぼ消失」と定義
sPGAで「消失」又は「ほぼ消失」を達成した患者の割合(ITT集団)
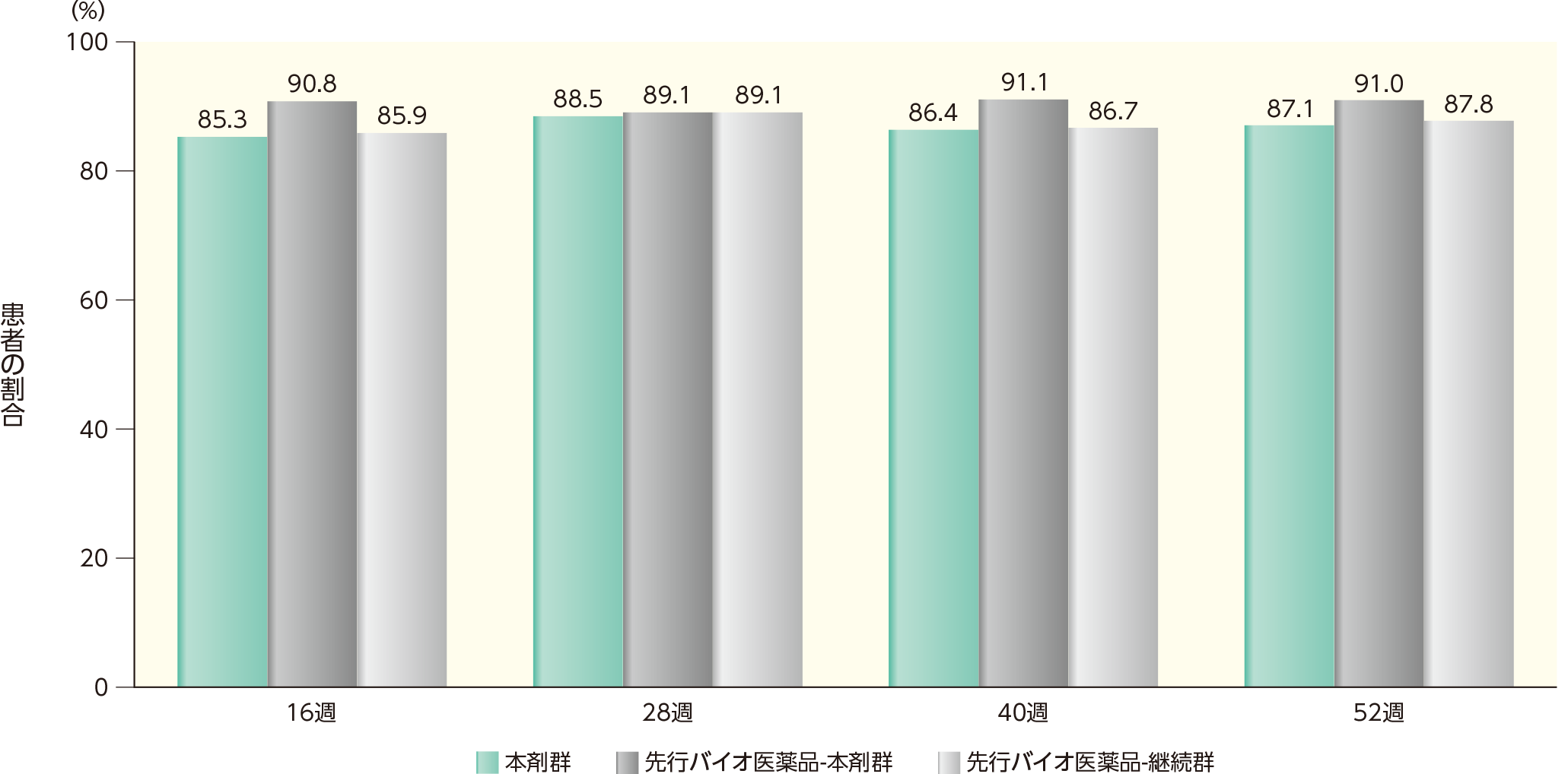
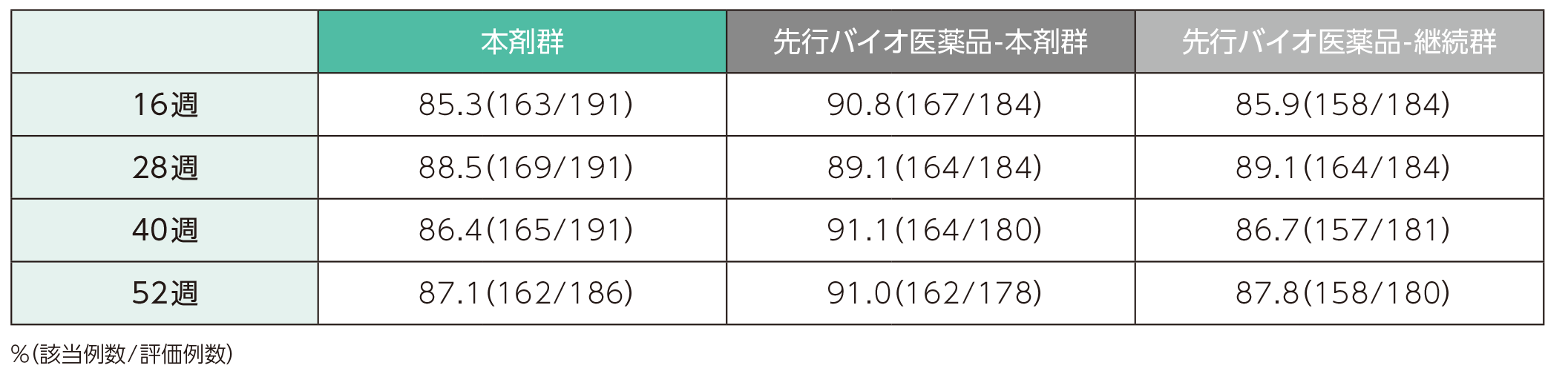
(2)16、28、40、52週時点にsPGAで「消失」又は「ほぼ消失」を達成した患者の割合ステージ2
16週時点において、sPGAで「消失」又は「ほぼ消失」を達成した患者の割合は、本剤群で85.3%、先行バイオ医薬品-本剤群で90.8%、先行バイオ医薬品-継続群で85.9%であった。各群において、52週時点までにsPGAで「消失」又は「ほぼ消失」を達成した患者の割合は以下のとおりであった。
*sPGAスコアが0の場合を「消失」、1の場合を「ほぼ消失」と定義
sPGAで「消失」又は「ほぼ消失」を達成した患者の割合(ITT集団)
【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の用法及び用量】
6.
用法及び用量 通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
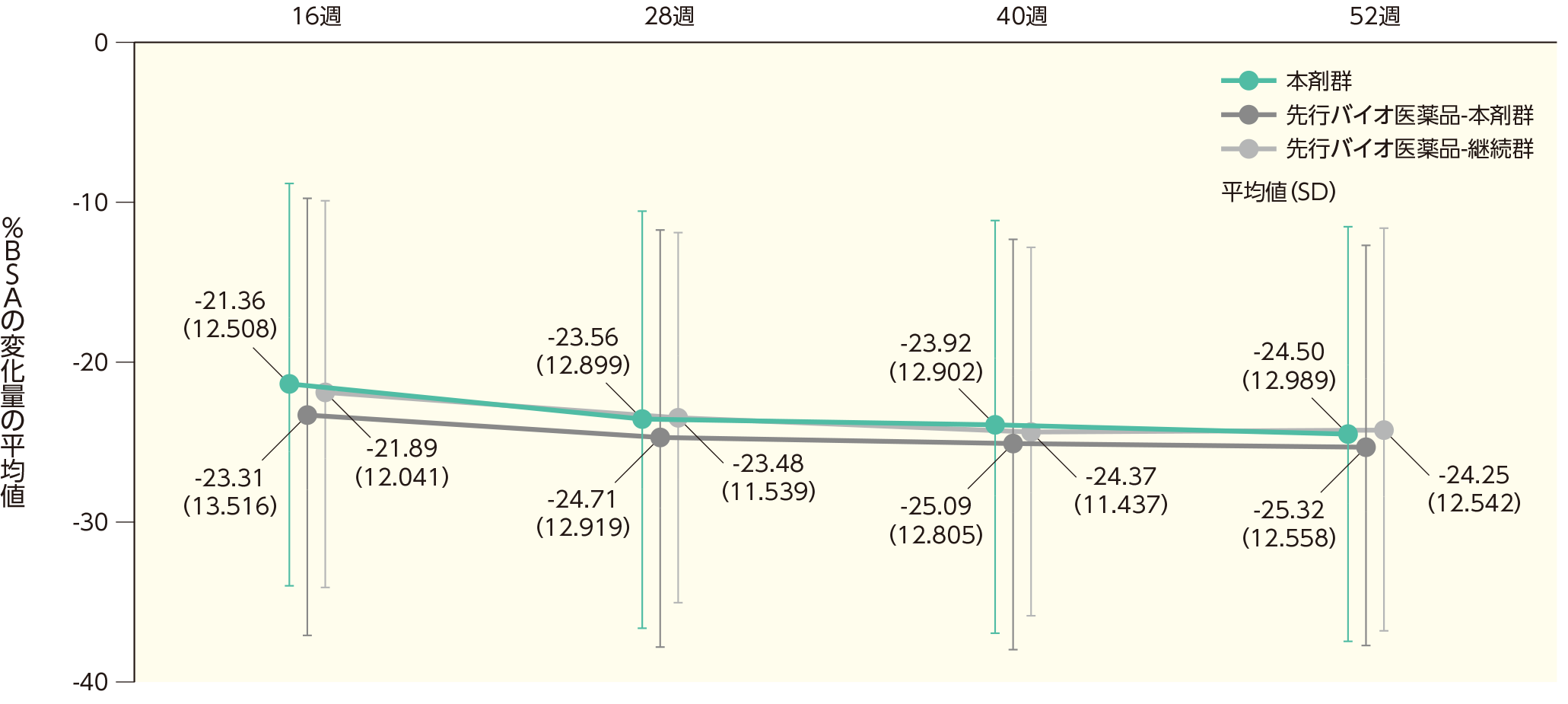
ベースラインから4、8、12、16、28、40、52週時点までの体表面積に占める乾癬病変の割合(%BSA)の変化(副次評価項目)
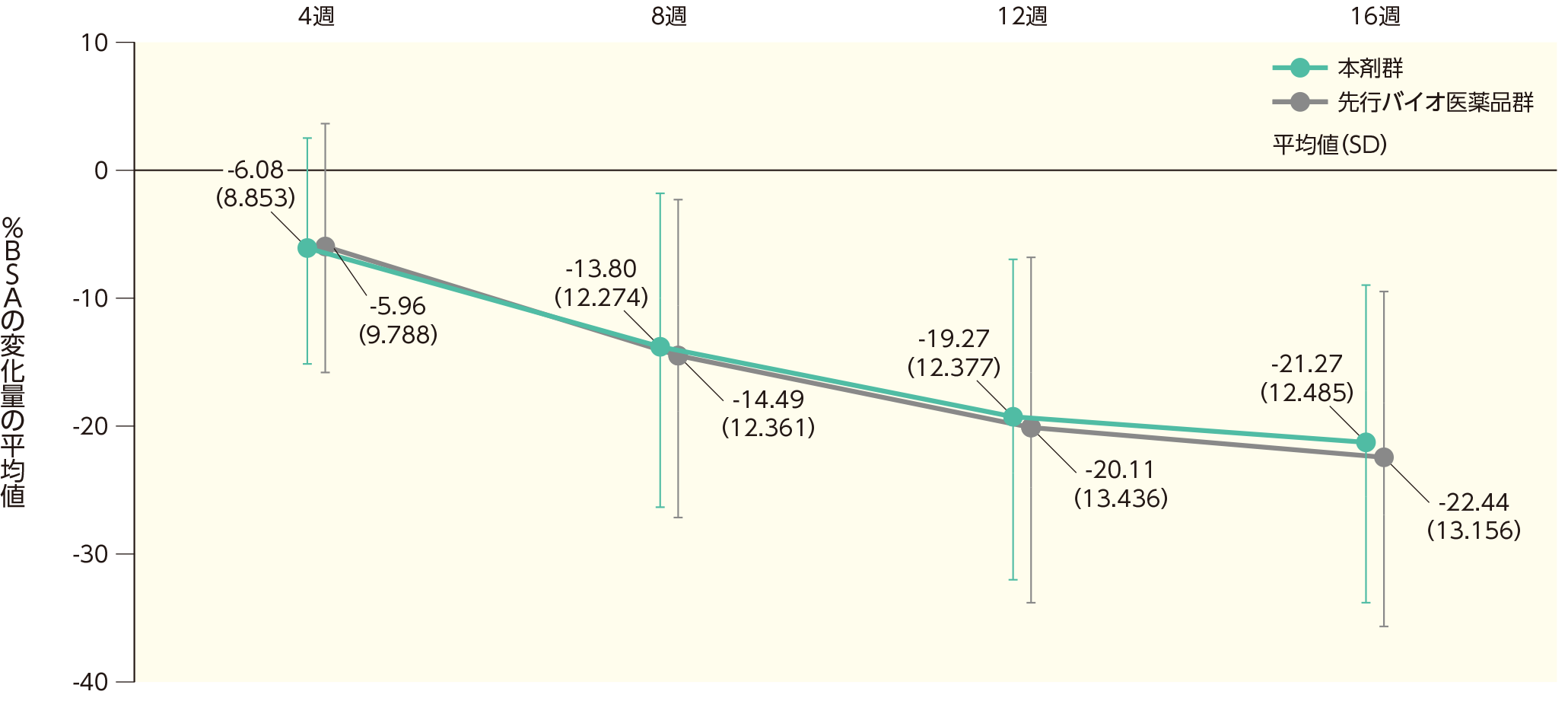
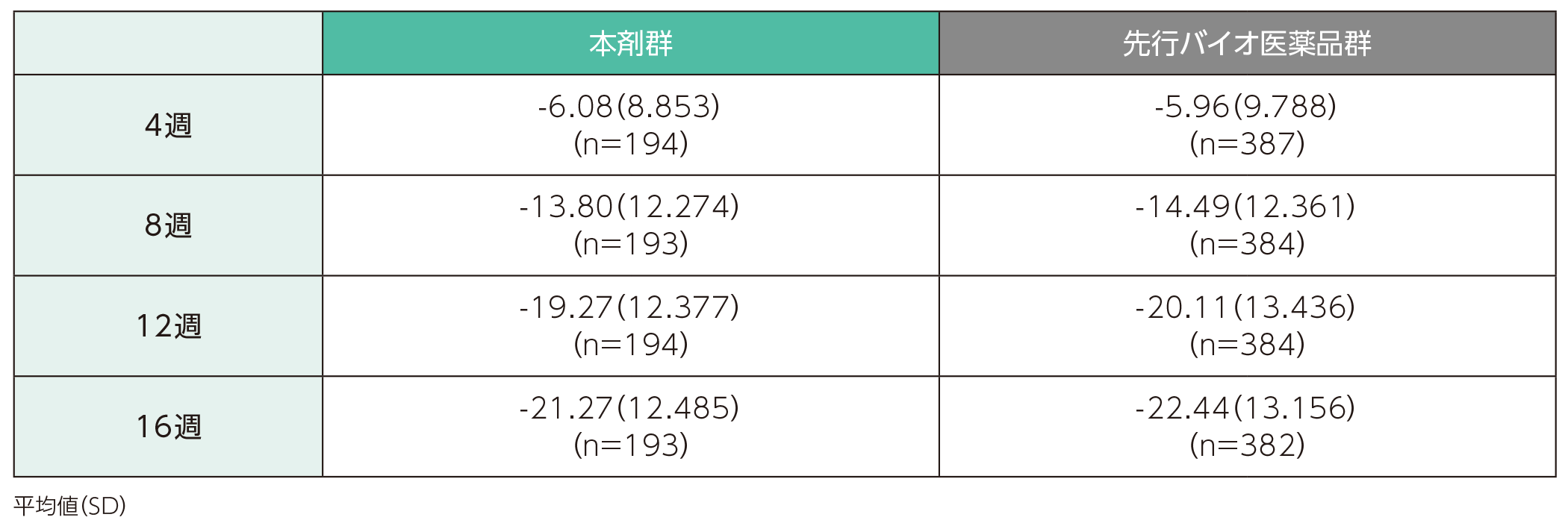
(1)ベースラインから4、8、12、16週時点までの%BSAの変化ステージ1
ベースラインから4週時点までの%BSAの変化量の平均値(SD)は、本剤群で-6.08(8.853)、先行バイオ医薬品群で-5.96(9.788)であった。各群の16週時点までの%BSAの変化量の平均値(SD)は以下のとおりであった。
%BSAのベースラインからの変化量(ITT集団)
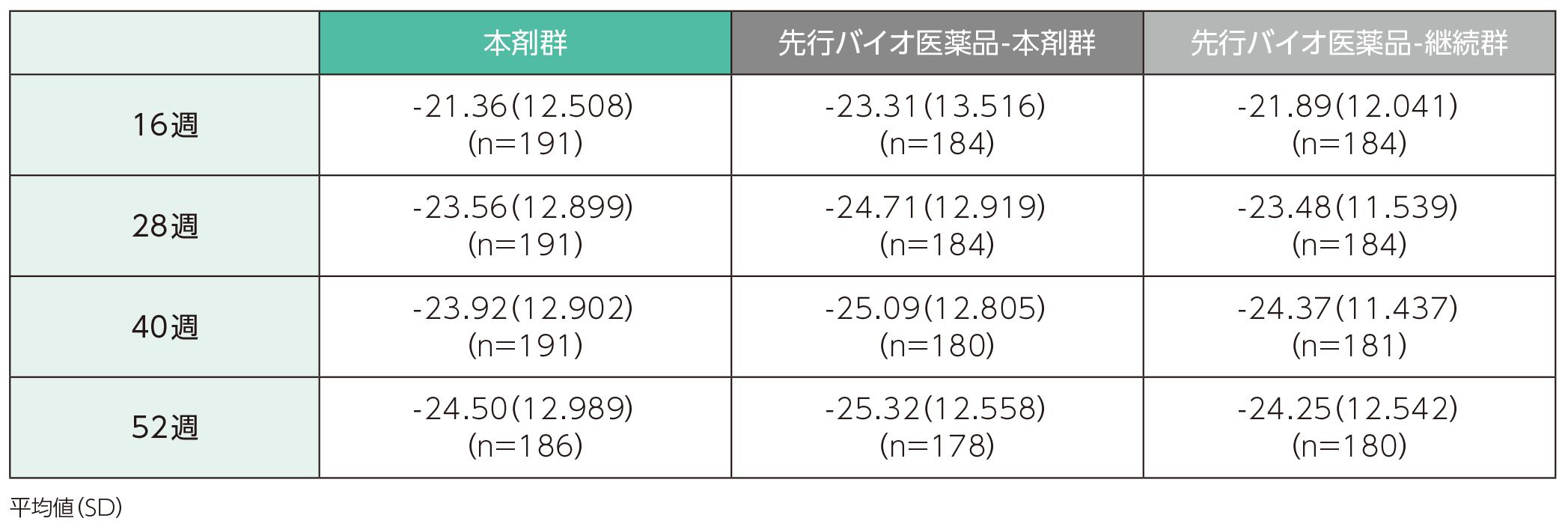
(2)ベースラインから16、28、40、52週時点までの%BSAの変化ステージ2
ベースラインから16週時点までの%BSAの変化量の平均値(SD)は、本剤群で-21.36(12.508)、先行バイオ医薬品-本剤群で-23.31(13.516)、先行バイオ医薬品-継続群で-21.89(12.041)であった。各群の52週時点までの%BSAの変化量の平均値(SD)は以下のとおりであった。
%BSAのベースラインからの変化量(ITT集団)
【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の用法及び用量】
6.
用法及び用量 通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
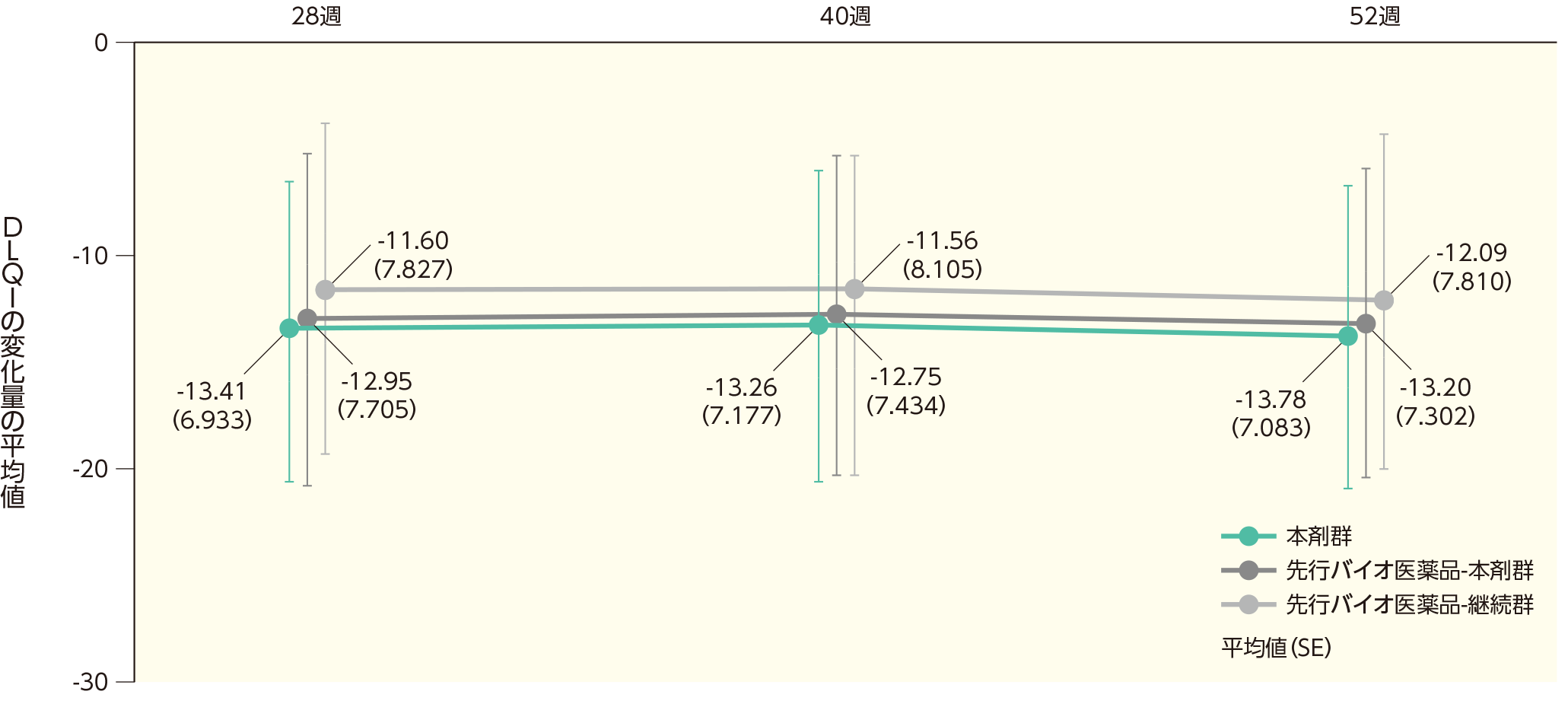
ベースラインから12、28、40、52週時点までの皮膚科関連QOL評価指標(DLQI)スコアの変化(副次評価項目)
(1)ベースラインから12週時点までのDLQIスコアの変化ステージ1
ベースラインから12週時点までのDLQIスコアの変化量の平均値(SD)は、本剤群で-12.48(7.141)、先行バイオ医薬品群で-11.41(7.928)であった。
DLQIスコアのベースラインからの変化量(ITT集団)
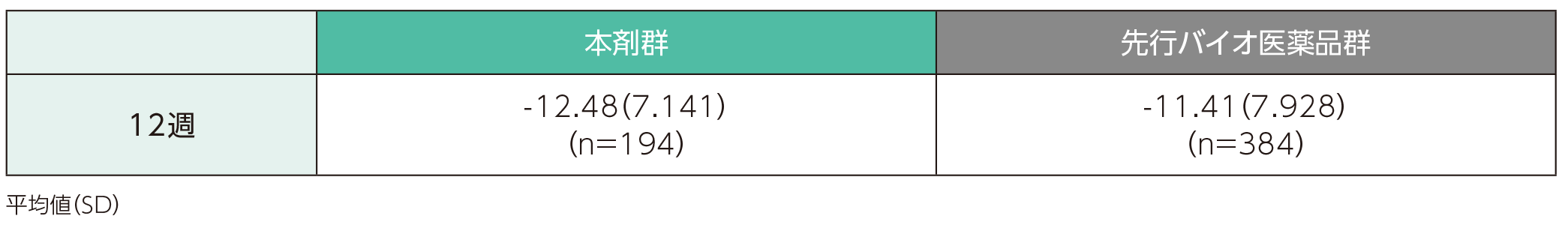
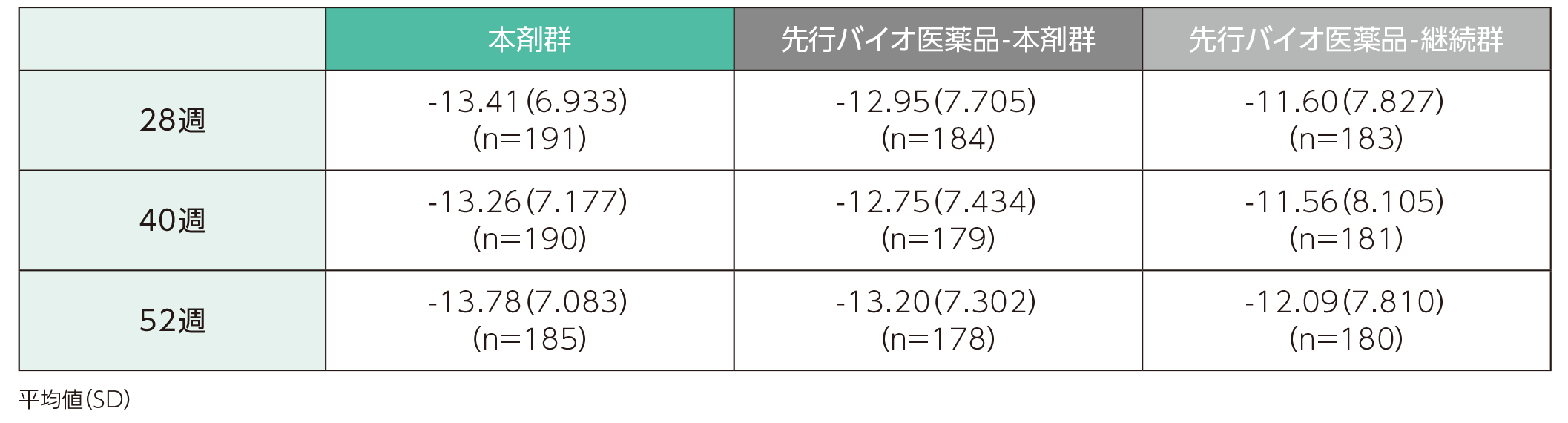
(2)ベースラインから28、40、52週時点までのDLQIスコアの変化ステージ2
ベースラインから28週時点までのDLQIスコアの変化量の平均値(SD)は、本剤群で-13.41(6.933)、先行バイオ医薬品-本剤群で-12.95(7.705)、先行バイオ医薬品-継続群で-11.60(7.827)であった。各群の52週時点までのDLQIスコアの変化量の平均値は以下のとおりであった。
DLQIスコアのベースラインからの変化量(ITT集団)
安全性
(1)16週時点までの安全性ステージ1
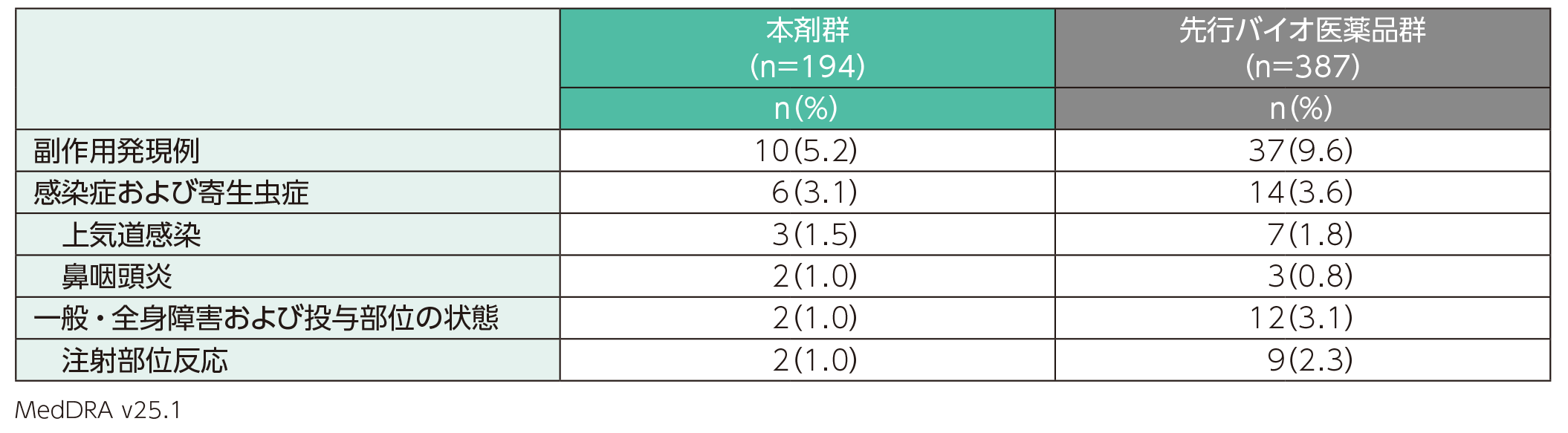
主な副作用
副作用は、本剤群では194例中10例(5.2%)、先行バイオ医薬品群では387例中37例(9.6%)に認められた。主な副作用(1%以上に発現)は、本剤群では上気道感染3例(1.5%)、鼻咽頭炎、注射部位反応各2例(1.0%)、先行バイオ医薬品群では注射部位反応9例(2.3%)、上気道感染7例(1.8%)であった。
重篤な副作用、投与中止に至った副作用、死亡に至った副作用
重篤な副作用、投与中止に至った副作用、死亡に至った副作用は、いずれも本剤群及び先行バイオ医薬品群では認められなかった。
(2)16週から28週時点までの安全性ステージ2
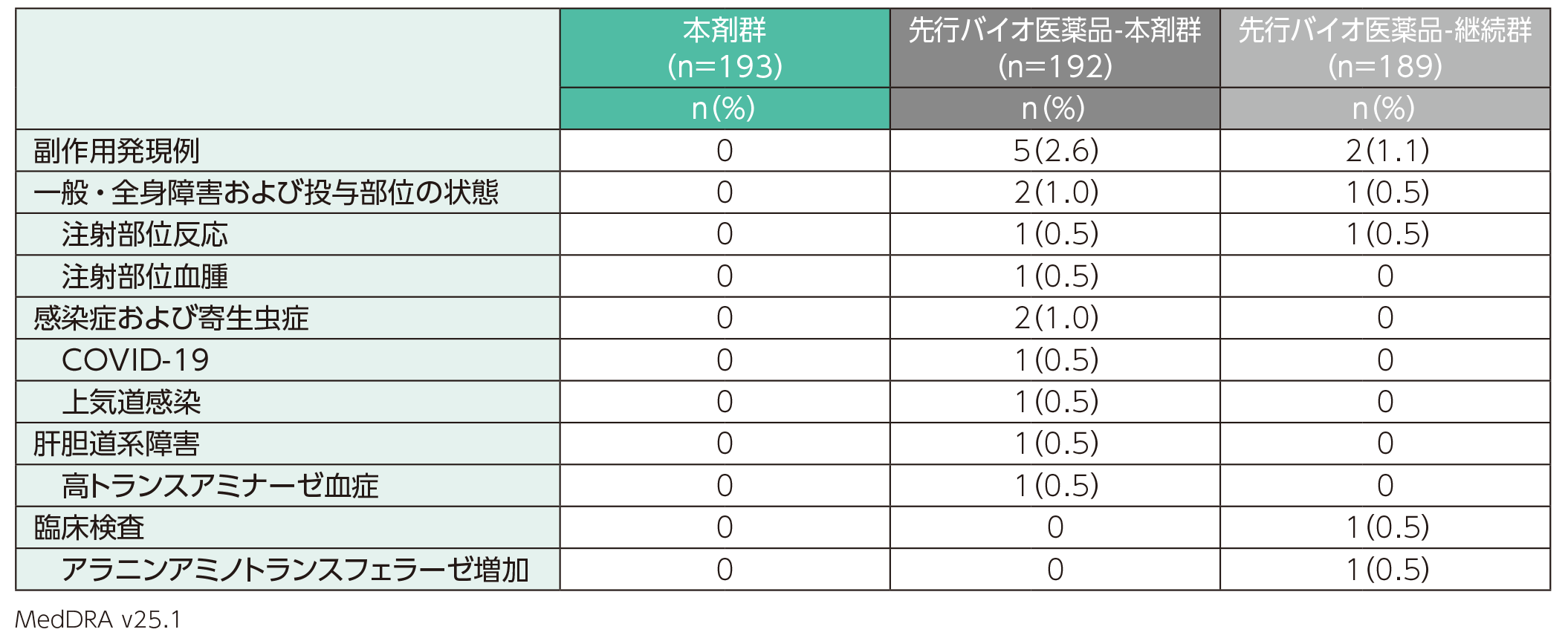
主な副作用
副作用は、先行バイオ医薬品-本剤群では192例中5例(2.6%)、先行バイオ医薬品-継続群では189例中2例(1.1%)に認められた。本剤群193例では認められなかった。副作用の内訳は、先行バイオ医薬品-本剤群では注射部位反応、注射部位血腫、COVID-19、上気道感染、高トランスアミナーゼ血症各1例(0.5%)、先行バイオ医薬品-継続群では注射部位反応、アラニンアミノトランスフェラーゼ増加各1例(0.5%)であった。
副作用(安全性解析対象集団)
重篤な副作用、投与中止に至った副作用、死亡に至った副作用
重篤な副作用、投与中止に至った副作用、死亡に至った副作用は、いずれも本剤群、先行バイオ医薬品-本剤群及び先行バイオ医薬品-継続群では認められなかった。
(3)28週から52週時点までの安全性ステージ2
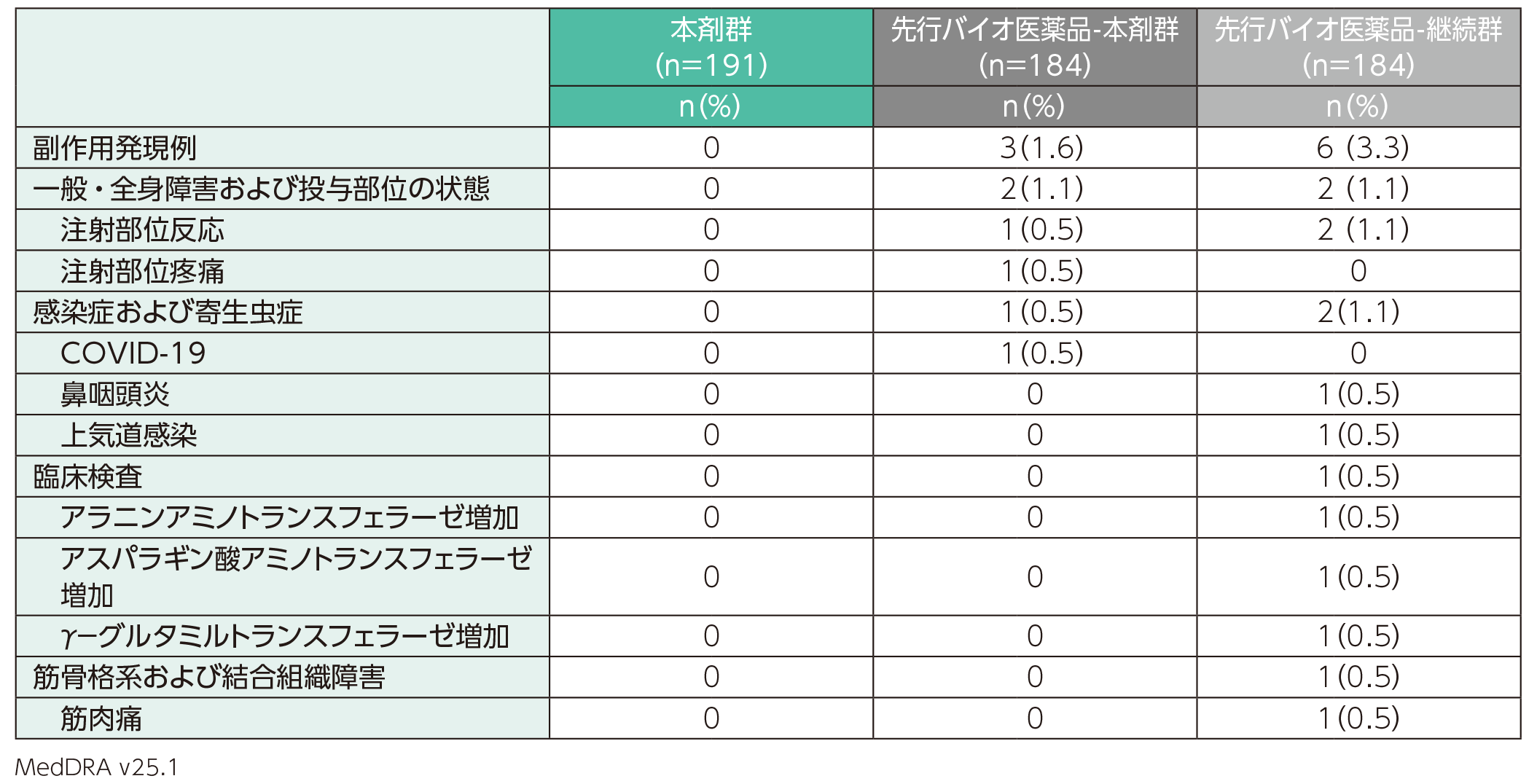
主な副作用
副作用は、先行バイオ医薬品-本剤群では184例中3例(1.6%)、先行バイオ医薬品-継続群では184例中6例(3.3%)に認められた。本剤群191例では認められなかった。副作用の内訳は、先行バイオ医薬品-本剤群では注射部位反応、注射部位疼痛、COVID-19各1例(0.5%)、先行バイオ医薬品-継続群では注射部位反応2例(1.1%)、鼻咽頭炎、上気道感染、アラニンアミノトランスフェラーゼ増加、アスパラギン酸アミノトランスフェラーゼ増加、γ-グルタミルトランスフェラーゼ増加、筋肉痛各1例(0.5%)であった。
副作用(安全性解析対象集団)
重篤な副作用、投与中止に至った副作用、死亡に至った副作用
重篤な副作用、投与中止に至った副作用、死亡に至った副作用は、いずれも本剤群、先行バイオ医薬品-本剤群及び先行バイオ医薬品-継続群では認められなかった。
【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の用法及び用量】
6.
用法及び用量 通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
4、12、16、28、40、52週時点に抗薬物抗体(ADA)及び中和抗体(nAb)が認められた患者の割合(副次評価項目)
(1)4、12、16週時点にADA及びnAbが認められた患者の割合 ステージ1
16週までのいずれかの時点でADAが認められた患者の割合は、本剤群では28.4%、先行バイオ医薬品群では54.5%であった。このうち、nAbが認められた割合は、本剤群では27.3%、先行バイオ医薬品群では32.2%であった。各群の16週時点までの各時点でADA、nAbが認められた患者の割合は以下のとおりであった。
ADAが認められた患者の割合(安全性解析対象集団)
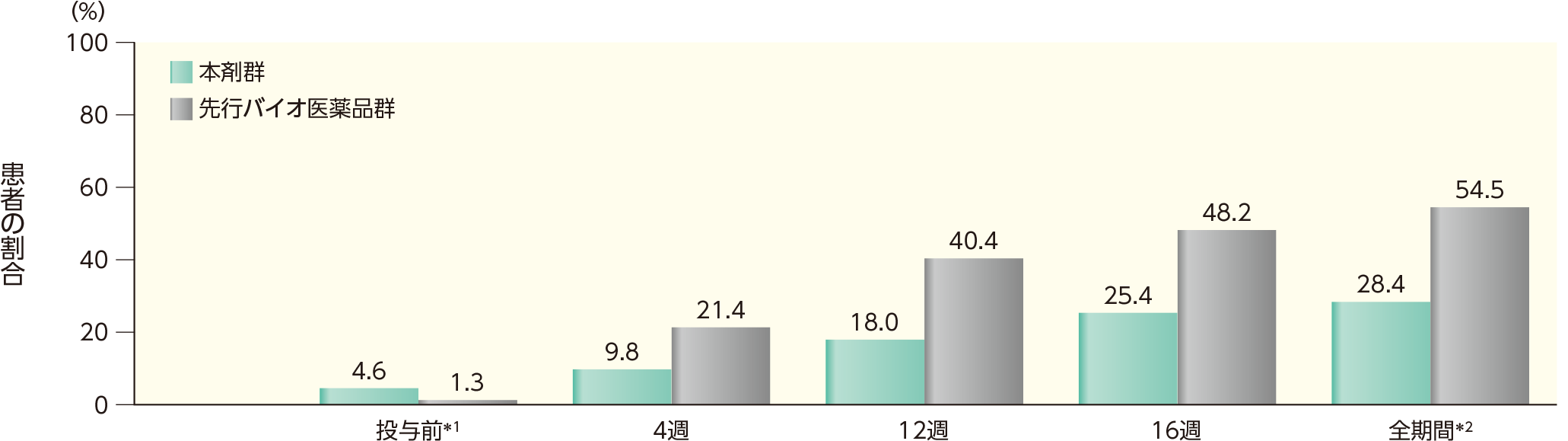

表中の数値はADA陽性例数/評価例数(発現率)を示す。
*1 ベースライン
*2 16週時点の投与前のいずれかの来院時にADAが認められた患者
nAbが認められた患者の割合(安全性解析対象集団)
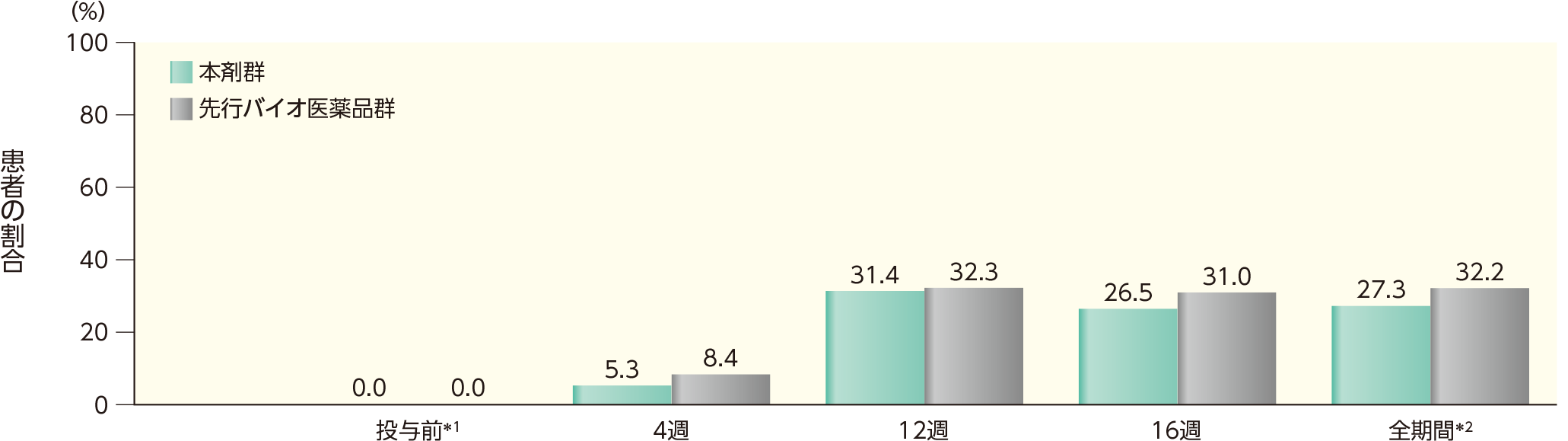

表中の数値はnAb陽性例数/ADA陽性例数(発現率)を示す。
*1 ベースライン
*2 16週時点の投与前のいずれかの来院時にnAbが認められた患者
【ウステキヌマブBS皮下注45mgシリンジ「F」・90mgシリンジ「F」の用法及び用量】
6.
用法及び用量 通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
(2)16、28、40、52週時点にADA及びnAbが認められた患者の割合ステージ2
52週時点までのいずれかの時点でADAが認められた患者の割合は、本剤群では38.7%、先行バイオ医薬品-本剤群では64.1%、先行バイオ医薬品-継続群では58.2%であった。このうち、nAbが認められた割合は、本剤群では32.4%、先行バイオ医薬品-本剤群では36.4%、先行バイオ医薬品-継続群では28.0%であった。また、各群の16週以降52週時点までの各時点でADA、nAbが認められた患者の割合は以下のとおりであった。
ADAが認められた患者の割合(安全性解析対象集団)
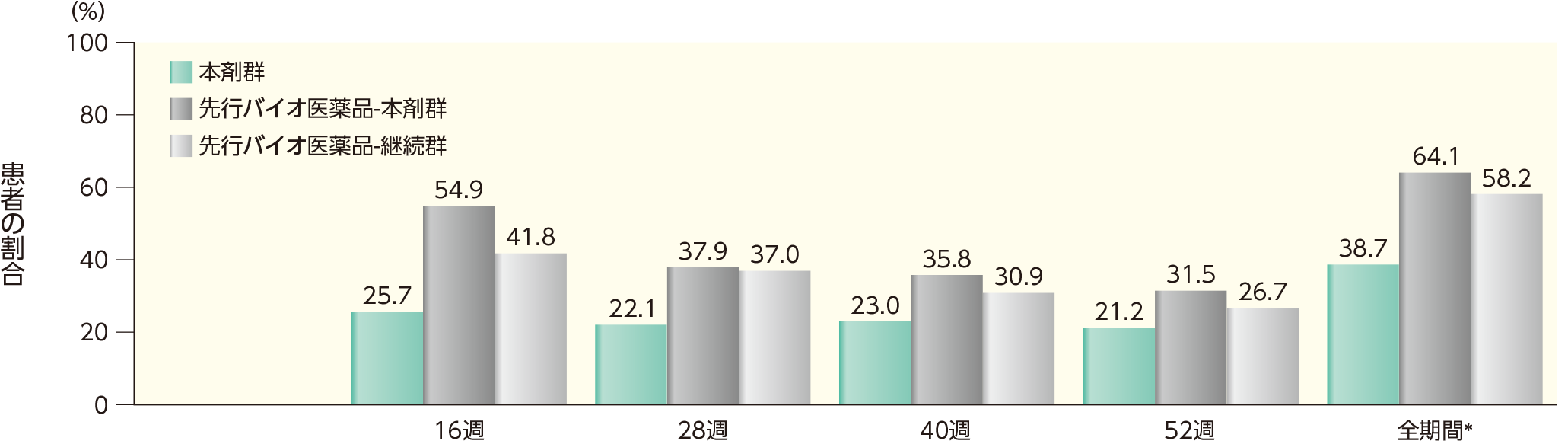
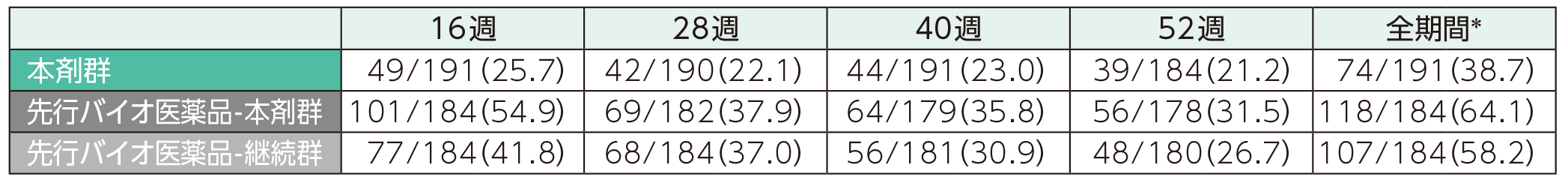
表中の数値はADA陽性例数/評価例数(発現率)を示す。
*52週時点までのいずれかの来院時にADAが認められた患者
nAbが認められた患者の割合(安全性解析対象集団)
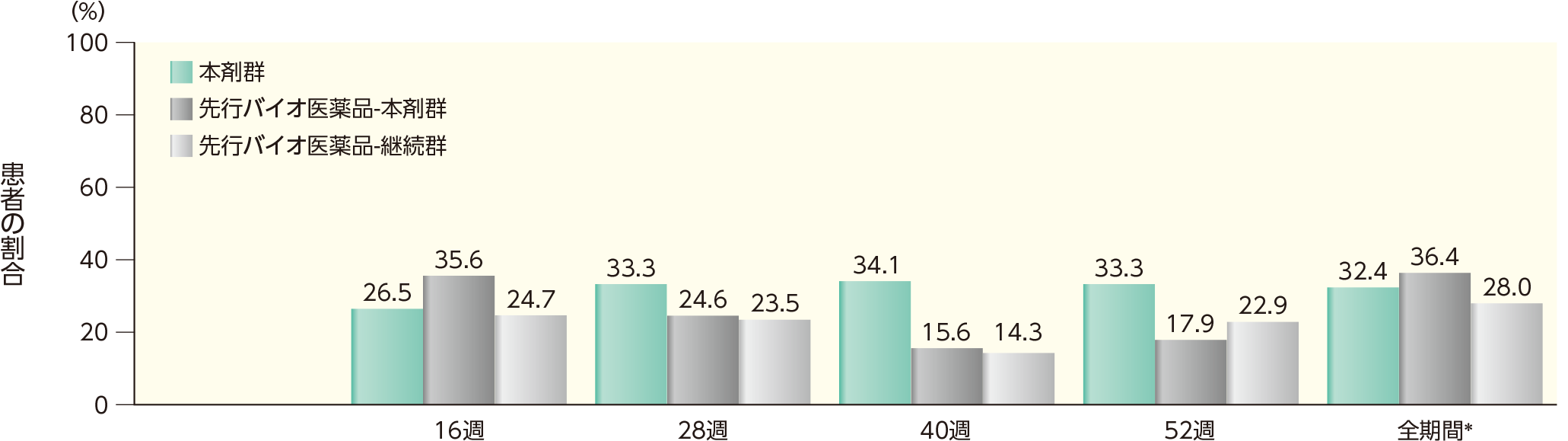
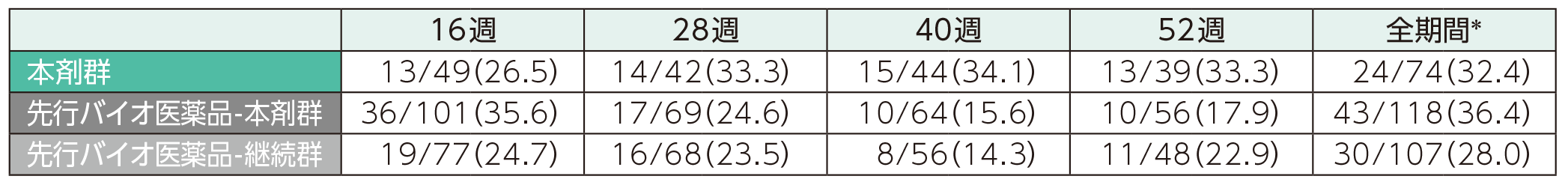
表中の数値はnAb陽性例数/ADA陽性例数(発現率)を示す。
*52週時点までのいずれかの来院時にnAbが認められた患者
1)社内資料: 海外第III相臨床試験成績(AVT04-GL-301)[承認時評価資料]
2)Feldman SR, et al.: Expert Opin Biol Ther. 2023; 23(8): 759-771
