
起こりうる副作用とその対策
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行ってください。
(1)重大な副作用
1)アナフィラキシー(発現頻度:不明※)
※先行バイオ医薬品(ステラーラ®)の臨床試験結果に基づく副作用発現頻度
アナフィラキシー(発疹、蕁麻疹、血管性浮腫等)があらわれることがあります
本剤投与時に観察すること2)重篤な感染症(発現頻度:1~5%未満※)
※先行バイオ医薬品(ステラーラ®)の臨床試験結果に基づく副作用発現頻度
ウイルス、細菌あるいは真菌による重篤な感染症(蜂巣炎、憩室炎、骨髄炎、胃腸炎、肺炎及び尿路感染等)があらわれることがあります。重篤な感染症が発現した場合には、感染が回復するまで本剤の投与をしないでください。
代表的な症状
- 発熱、咳、呼吸困難などの症状があらわれます。
対処方法
- 発熱、咳、呼吸困難などの症状が出現した場合は、フローチャート(下図)に従って診断を行い、各感染症に応じた抗菌剤治療を行ってください。
- 本剤の投与は中止し、感染症が消失するまで再投与しないでください。
- 高齢者、肺疾患を有する患者さん、ステロイドを服用している患者さんなど、重篤な感染症を発症するリスクが高いと考えられる患者さんには、ST合剤などの積極的な予防投与を考慮してください。
1.
警告(抜粋)
1.2
重篤な感染症
ウイルス、細菌及び真菌による重篤な感染症が報告されているため、十分な観察を行うなど感染症の発症に注意すること。[2.1、8.1、9.1.1、11.1.2 参照]
投与中の注意事項
- 十分な観察及び問診を行い、投与中は感染症の徴候に十分注意してください。
生物学的製剤、JAK阻害薬投与中における発熱、咳、呼吸困難に対するフローチャート
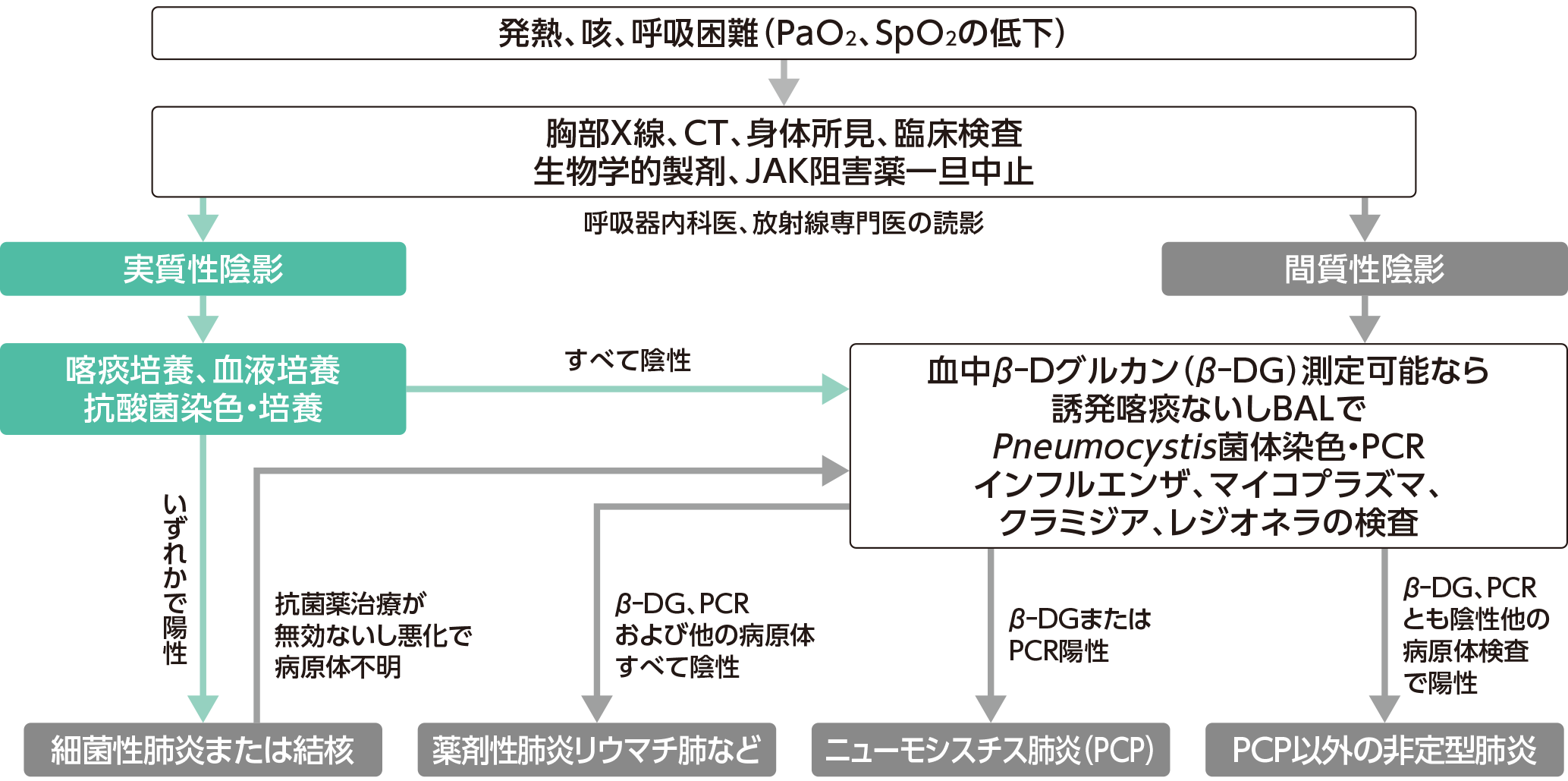
日本リウマチ学会: 関節リウマチ(RA)に対するTNF阻害薬使用の手引き(2024年7月7日改訂版)
https://www.ryumachi-jp.com/pdf/tebiki_tnf_240710.pdf
詳細は日本リウマチ学会ホームページをご参照ください。
3)結核(発現頻度:不明※)
※先行バイオ医薬品(ステラーラ®)の臨床試験結果に基づく副作用発現頻度
胸部画像検査等の適切な検査を定期的に行うなど、結核の徴候及び症状*に注意し、結核の症状が疑われる場合には、結核の診療経験のある医師に相談してください。
また、患者さんに対しても結核の症状が疑われる場合には、速やかに担当医師に連絡するようご指導ください。
*結核初期の臨床症状は、全くの無症状から重度の呼吸不全までさまざまですが、全身症状としては、発熱・全身倦怠感・体重減少などを呈し、呼吸器症状としては、咳・喀痰・血痰・胸痛・呼吸困難などを呈します。
結核が発現又は再活性化する可能性があります。
代表的な症状
- 結核の代表的な症状として、2週間以上続く咳、痰、微熱が挙げられます。食欲不振、倦怠感、急激な体重の減少などの症状もみられます。進行すると、血痰、喀血、呼吸困難などの症状を伴うこともあります。
対処方法
- 結核の疑いのある患者さんには、本剤投与前にあらかじめ結核に対する治療を行ってください。本剤投与開始3週間前より、イソニアジドを原則300mg/日*、通常は6ヵ月間、糖尿病の合併や免疫抑制状態が想定される場合は9ヵ月間経口投与してください。
- 結核に対する治療については、呼吸器内科医、放射線専門医、感染症専門医などの専門医と連携しながら行ってください。
投与中の注意事項
- 結核感染の有無を確認するため、必要に応じて定期的(6ヵ月ごと)に胸部X線検査などを行ってください。
*イソニアジドの用法及び用量は以下のとおりです。
用法及び用量:通常成人は、イソニアジドとして1日量200~500mg(4~10mg/kg)を筋肉内又は静脈内注射する。髄腔内、胸腔内注入又は局所分注の場合には1回50~200mgを使用する。年齢、症状により適宜増減する。なお、他の抗結核薬と併用することが望ましい。
1.
警告(抜粋)
1.3
結核等の感染症について診療経験を有する内科等の医師と十分な連携をとり使用すること。[2.2、8.2、9.1.2、11.1.3 参照]
4)間質性肺炎(発現頻度:不明※)
※先行バイオ医薬品(ステラーラ®)の臨床試験結果に基づく副作用発現頻度
本剤投与例において、間質性肺炎の発現が報告されています。また、間質性肺炎の既往を有する患者さんにおける間質性肺炎の再燃が報告されています。
対処方法
- 間質性肺炎が疑われた場合には投与を中止し、副腎皮質ホルモン剤の投与等の適切な処置を行ってください。
投与中の注意事項
- 咳嗽、呼吸困難、発熱、肺音の異常(捻髪音)等が認められた場合には、速やかに胸部X線、胸部CT、血清マーカー等の検査を実施してください。
- 高齢者、間質性肺炎の既往例又は合併症を有する患者さんにおいては、定期的に問診を行うなど、注意してください。
(2)悪性腫瘍
本剤はIL−12/23の作用を選択的に抑制する薬剤であり、悪性腫瘍発現の可能性があり、皮膚及び皮膚以外の悪性腫瘍の発現が報告されています。本剤との因果関係は明確ではありませんが、悪性腫瘍の発現には注意してください。
海外の尋常性乾癬、乾癬性関節炎、クローン病(承認外効能・効果)、潰瘍性大腸炎(承認外効能・効果)を対象とした臨床試験(第II相及び第III相試験)において、プラセボ対照期間の非黒色腫皮膚癌を除く悪性腫瘍の発現頻度は、本剤投与群が0.11/100人年(1例/929人年)、プラセボ投与群が0.23/100人年(1例/434人年)でした。非黒色腫皮膚癌の発現頻度は、本剤投与群が0.43/100人年(4例/929人年)、プラセボ投与群が0.46/100人年(2例/433人年)でした。また、対照及び非対照期間において、6710名(15205人年)に本剤が投与されました。その追跡調査中央値は1.2年で、尋常性乾癬を対象とした臨床試験では3.2年、乾癬性関節炎を対象とした臨床試験では1.5年、クローン病注)を対象とした臨床試験では0.6年、潰瘍性大腸炎注)を対象とした臨床試験では2.3年でした。非黒色腫皮膚癌を除く悪性腫瘍の発現頻度は、0.50/100人年(76例/15205人年)で、主なものは前立腺癌、黒色腫、結腸直腸癌、乳癌でした。本剤投与群で報告された悪性腫瘍の発現頻度は、一般人口で予測される発現頻度と同様でした(標準化発生比:0.94[95%信頼区間:0.73、1.18]年齢、性別、人種により補正)。非黒色腫皮膚癌の発現頻度は0.46/100人年(56例/11545人年)でした。皮膚基底細胞癌と皮膚有棘細胞癌の発現比率は3:1であり一般人口で予測される発現頻度と同様でした。
注)本剤の効能又は効果:既存治療で効果不十分な下記疾患 尋常性乾癬、乾癬性関節炎
対処方法
- 悪性腫瘍の発現が認められた場合は、本剤の投与を中止し、悪性腫瘍に対する治療を行ってください。必要に応じて専門医へ紹介してください。
投与中の注意事項
- 投与中は定期的ながん検診を受けるように患者さんにご指導ください。
1.
警告(抜粋)
1.1
本剤はIL−12/23の作用を選択的に抑制する薬剤であるため、感染のリスクを増大させる可能性がある。また、結核の既往歴を有する患者では結核を活動化させる可能性がある。また、本剤との関連性は明らかではないが、悪性腫瘍の発現が報告されている。本剤が疾病を完治させる薬剤でないことも含め、これらの情報を患者に十分説明し、患者が理解したことを確認した上で、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。また、本剤投与後に副作用が発現した場合には、主治医に連絡するよう患者に注意を与えること。[2.1、8.1-8.3、9.1.1-9.1.3、11.1.2、11.1.3、15.1.6参照]
4.
効能又は効果
○既存治療で効果不十分な下記疾患
尋常性乾癬、乾癬性関節炎
(3)副作用一覧
本試験には承認外の用法及び用量の成績が含まれているが、本剤と先行バイオ医薬品(EU)*との有効性の同等性を検討した承認時評価資料のため紹介している。
*先行バイオ医薬品(EU)は、欧州で承認されているステラーラ®〔ウステキヌマブ(遺伝子組換え)製剤〕を指す。
〈海外第III相臨床試験(AVT04-GL-301試験、海外データ)1,2)、※〉
1)社内資料:海外第III相臨床試験成績(AVT04-GL-301)[承認時評価資料]
2)Feldman SR, et al.: Expert Opin Biol Ther. 2023; 23(8): 759-771
(COI:本試験はAlvotech社の資金提供により実施された。著者の中にAlvotech社の社員が含まれる)
※試験概要はページ下部をご参照ください。
1)16週時点までの安全性 ステージ1
副作用は、本剤群では194例中10例(5.2%)、先行バイオ医薬品群では387例中37例(9.6%)に認められた。主な副作用(1%以上に発現)は、本剤群では上気道感染3例(1.5%)、鼻咽頭炎、注射部位反応各2例(1.0%)、先行バイオ医薬品群では注射部位反応9例(2.3%)、上気道感染7例(1.8%)であった。
副作用(安全性解析対象集団)
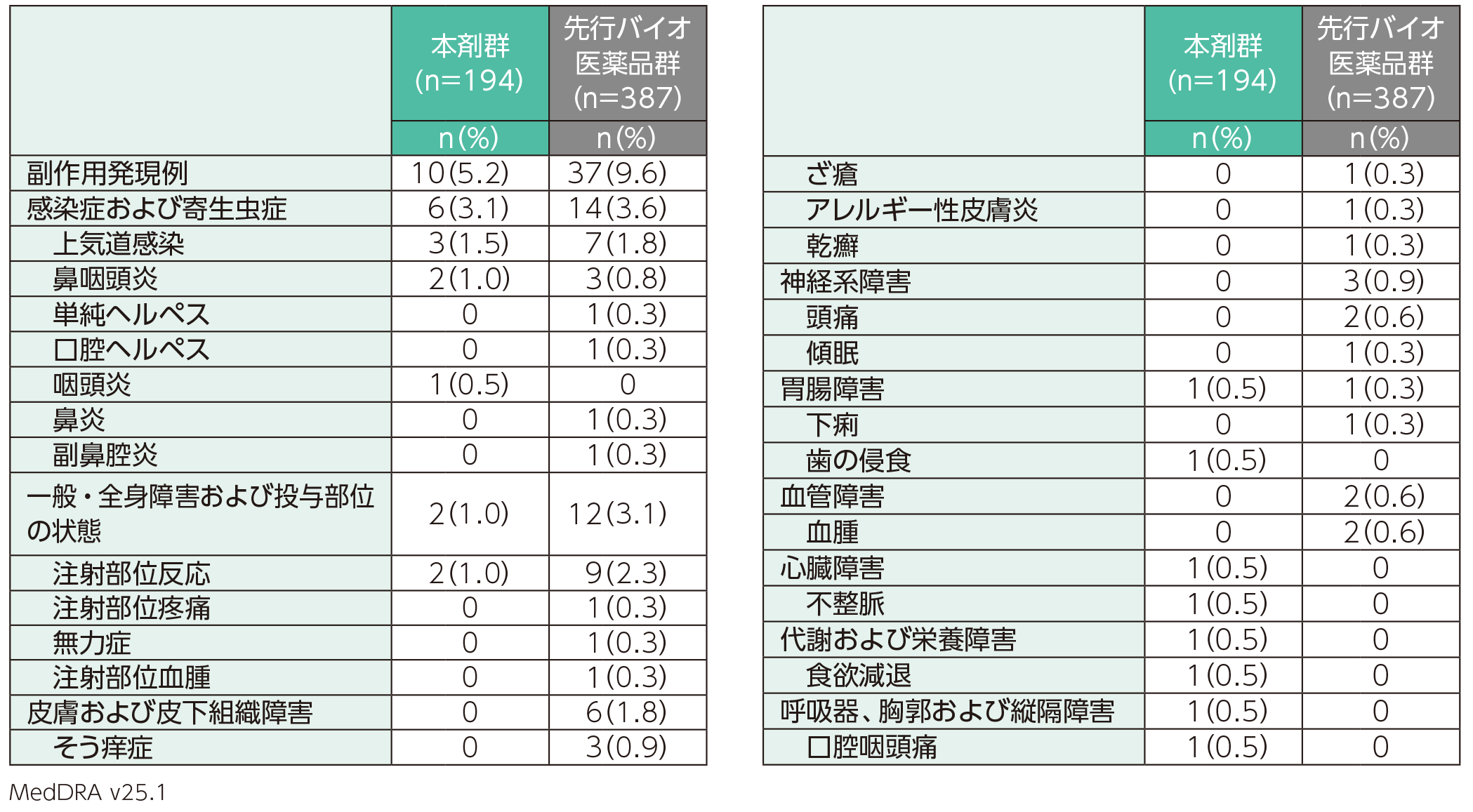
また、重篤な副作用、投与中止に至った副作用、死亡に至った副作用は、いずれも本剤群及び先行バイオ医薬品群では認められなかった。
6.
用法及び用量
通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
2)16週から28週時点までの安全性 ステージ2
副作用は、先行バイオ医薬品-本剤群では192例中5例(2.6%)、先行バイオ医薬品-継続群では189例中2例(1.1%)に認められた。本剤群193例では認められなかった。副作用の内訳は、先行バイオ医薬品-本剤群では注射部位反応、注射部位血腫、COVID-19、上気道感染、高トランスアミナーゼ血症各1例(0.5%)、先行バイオ医薬品-継続群では注射部位反応、アラニンアミノトランスフェラーゼ増加各1例(0.5%)であった。
副作用(安全性解析対象集団)
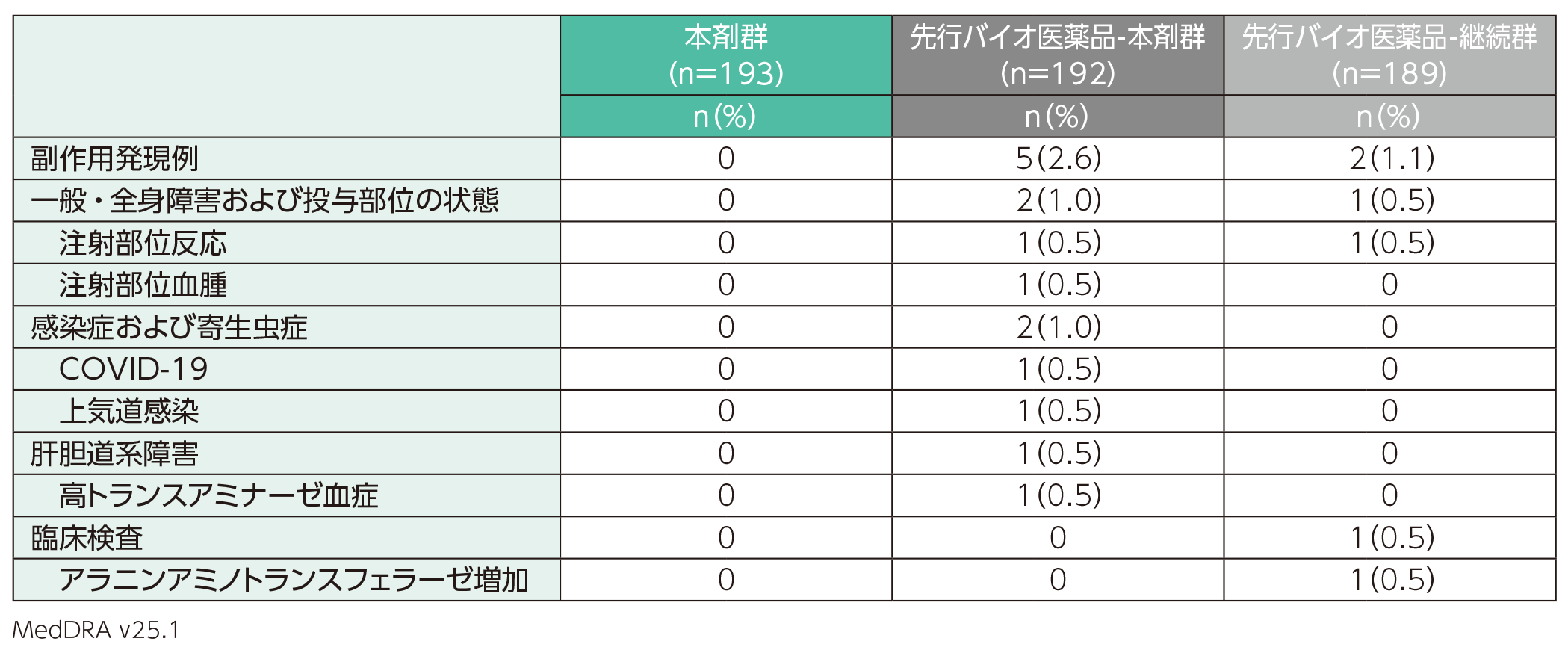
また、重篤な副作用、投与中止に至った副作用、死亡に至った副作用は、いずれも本剤群、先行バイオ医薬品-本剤群及び先行バイオ医薬品-継続群では認められなかった。
3)28週から52週時点までの安全性 ステージ2
副作用は、先行バイオ医薬品-本剤群では184例中3例(1.6%)、先行バイオ医薬品- 継続群では184例中6例(3.3%)に認められた。本剤群191例では認められなかった。副作用の内訳は、先行バイオ医薬品-本剤群では注射部位反応、注射部位疼痛、COVID-19各1例(0.5%)、先行バイオ医薬品-継続群では注射部位反応2例(1.1%)、鼻咽頭炎、上気道感染、アラニンアミノトランスフェラーゼ増加、アスパラギン酸アミノトランスフェラーゼ増加、γ−グルタミルトランスフェラーゼ増加、筋肉痛各1例(0.5%)であった。
副作用(安全性解析対象集団)
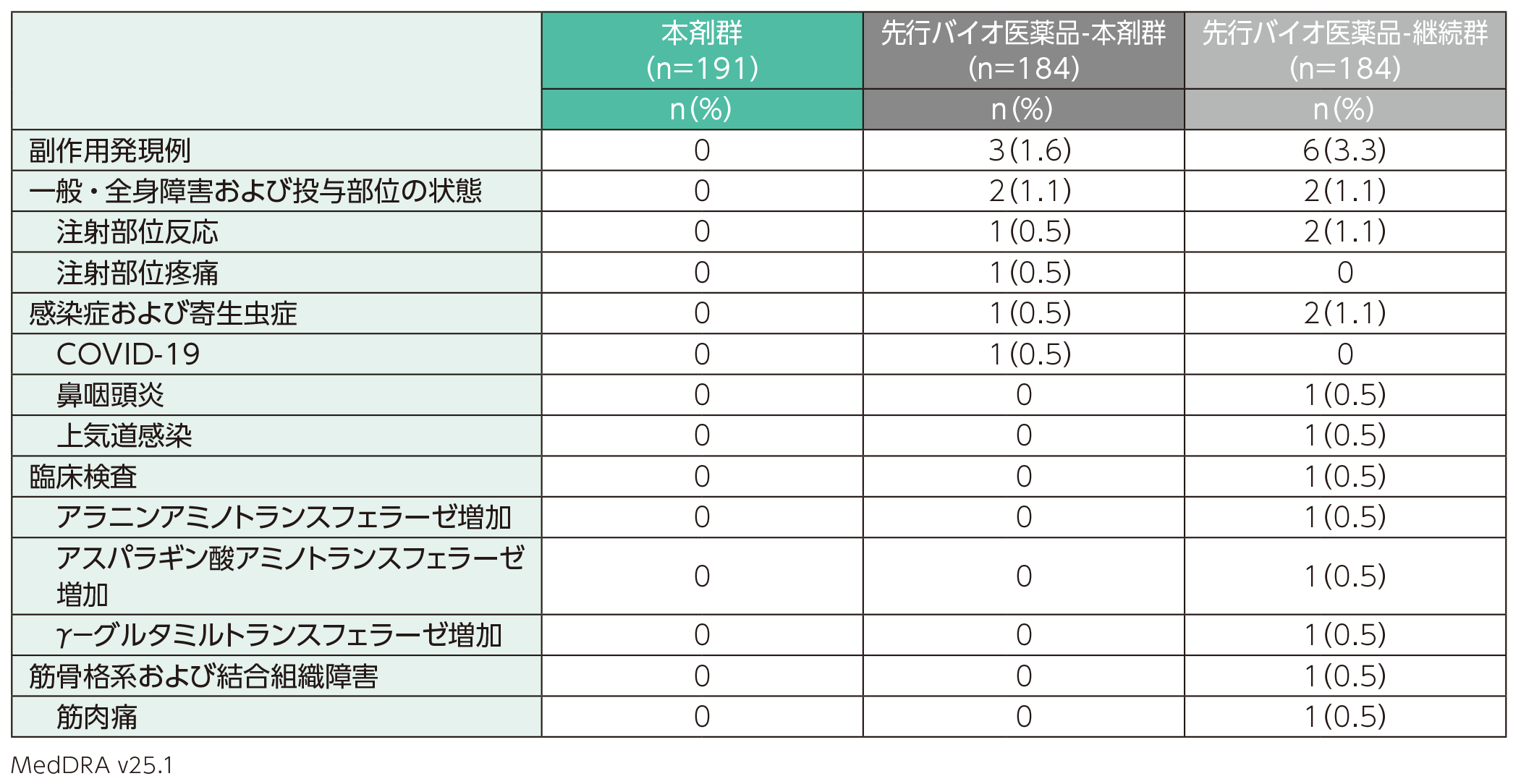
また、重篤な副作用、投与中止に至った副作用、死亡に至った副作用は、いずれも本剤群、先行バイオ医薬品-本剤群及び先行バイオ医薬品-継続群では認められなかった。
4)試験概要
| 目的 | 中等症又は重症の慢性尋常性乾癬患者を対象に、本剤と先行バイオ医薬品(EU)*1の有効性、安全性及び免疫原性を比較する。 |
|---|---|
| 試験デザイン | 無作為化、二重盲検、多施設共同試験 |
| 対象 | 中等症又は重症の慢性尋常性乾癬患者581例 |
| 試験方法 | 本試験はステージ1(初回投与から16週目まで)及びステージ2(16週目から52週目まで)で構成された。対象を、尋常性乾癬に対する生物学的製剤による治療歴の有無及び体重(80kg以下、80kg超-100kg以下、100kg超)を層別因子として層別し、本剤群又は先行バイオ医薬品群のいずれかに1:2の割合で無作為割り付けした。 本剤群:本剤の初回皮下投与*2を実施し、4週後に2回目の皮下投与*2を行った。その後は、以降40週まで12週ごとに皮下投与*2した。 先行バイオ医薬品群:ステージ1では、先行バイオ医薬品(EU)の初回皮下投与*2を実施し、4週後に2回目の皮下投与*2を行った。ステージ2では、先行バイオ医薬品-本剤群(本剤を皮下投与*2)又は先行バイオ医薬品-継続群〔先行バイオ医薬品(EU)を皮下投与*2〕に1:1の割合で無作為割り付けし、16、28、40週時点に皮下投与*2した。なお、ベースラインと比較して、28週時点の乾癬面積重症度指数(PASI)改善率が50%未満の場合は試験を中止した。 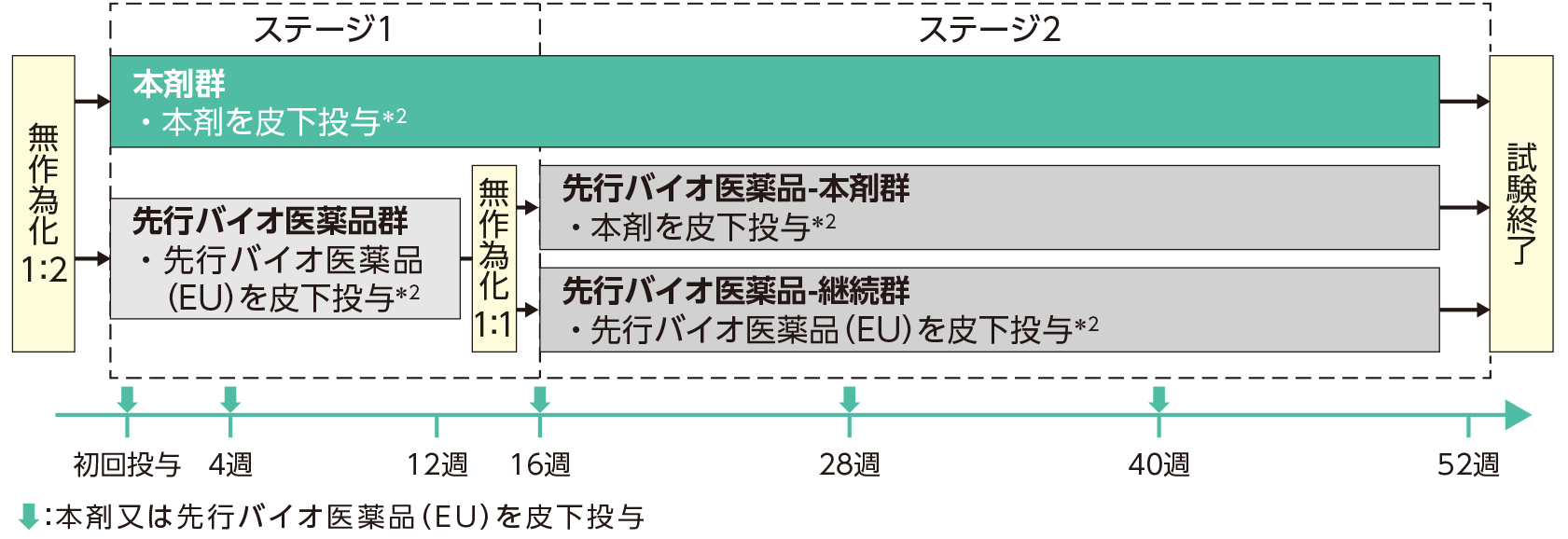 |
| 主要評価項目 | [有効性] ベースラインから12週時点までのPASI改善率[検証的解析項目] |
| 副次評価項目 | [有効性]
|
| 解析計画 | [有効性の解析] 副次評価項目:解析はITT集団*4を対象に、本剤群と先行バイオ医薬品群の有効性を解析し、記述的に要約した。ベースラインからのPASI改善率及びベースラインから12週時点までのPASIのAUECの解析には、主要評価項目の解析に用いたANCOVAモデルを適用した。なお、多重性の調整は実施しなかった。 [安全性の解析] [免疫原性解析] [PKの解析] |
| 試験期間 | 2021年6月3日(最初の患者の初回来院)~2022年10月11日(最後の患者の最終来院) |
*1 先行バイオ医薬品(EU)は、欧州で承認されているステラーラ®〔ウステキヌマブ(遺伝子組換え)製剤〕を指す。
*2 患者体重が100kg以下の場合は本剤又は先行バイオ医薬品(EU)45mg、100kg超の場合は本剤又は先行バイオ医薬品(EU)90mgを皮下投与※
※初回からウステキヌマブ90mg投与は国内未承認です。
*3 PPS集団:治験実施計画書からの逸脱がなく、投与期間を完了した患者
*4 ITT集団:無作為化後に割り付けられた治験薬を1回以上投与されたすべての患者
*5 各有害事象はMedDRA v25.1に基づく。
*6 安全性解析対象集団:無作為化後に割り付けられた治験薬を1回以上投与されたすべての患者
6.
用法及び用量
通常、成人にはウステキヌマブ(遺伝子組換え)[ウステキヌマブ後続1]として1回45mgを皮下投与する。初回投与及びその4週後に投与し、以降12週間隔で投与する。ただし、効果不十分な場合には1回90mgを投与することができる。
